摘要:近年来,微生物单细胞RNA测序(single-microbe RNA sequencing,smRNA-seq)技术得到了显著发展,特别是在细菌单细胞转录组分析领域,涌现了如microSPLiT[1]、PETRI-seq[2]、smRandom-seq[3] 等多种新型方法。然而,数据分析方面的进展相对滞后,现有smRNA-seq分析流程大多依赖于真核单细胞RNA测序(single-cell RNA sequencing,scRNA-seq)的框架,并对参数和阈值进行适应性修改。为解决这一问题,近年来出现了几个针对smRNA-seq的专门分析框架,其中包括smRandom-seq2[4]、MscT[5] 和mKmer[6]。本文重点介绍了smRNA-seq的主要数据分析流程和工具,并探讨了当前几种代表性分析框架的创新点。
关键词: smRNA-seq, 单细胞转录组学, 数据分析框架, 多组学整合, 泛基因组分析
背景
微生物单细胞转录组与真核细胞转录组在生物学上存在显著差异。微生物多为原核生物,缺乏细胞核与膜性细胞器,其基因组通常无内含子、以多基因操纵子形式组织,转录与翻译同时在细胞质中进行,mRNA无poly(A)尾且半衰期极短,导致基因表达呈现强烈的瞬态和高噪声特征;而真核细胞具核膜和复杂的转录后加工机制,mRNA稳定性高,表达调控层级丰富。此外,微生物细胞体积小、rRNA含量高,实验上裂解和RNA捕获更具挑战。其表达异质性更多反映代谢状态和环境适应差异,而非发育或组织特异性,因此分析重点常聚焦于功能通路和调控模块层面的表达模式,生物学上的差异也导致测序技术和分析工具选择的不同。近五年来,微生物单细胞RNA测序技术发展迅速,先后出现了多种细菌scRNA-seq方法:基于平板的microSPLiT[1]、PETRI-seq[2]、MATQ-seq[7],基于探针的 par-seqFISH[8]、ProBac-seq[9],以及基于液滴的BacDrop[10]、smRandom-seq[3])。相比之下,数据分析的发展相对滞后,目前大多数微生物单细胞转录组(smRNA-seq)数据分析仍主要沿用真核scRNA-seq的框架和工具,仅在参数和阈值上进行适配性修改。近年陆续出现了一些专门面向smRNA-seq的计算框架,例如:smRandom-seq2[4] 首次实现了复杂微生物群落的多物种单细胞分析;MscT[5] 建立了从测序、参考数据库构建到计算分析的完整流程,其中基于泛基因组的注释方法为smRNA-seq提供了有效的解决方案;mKmer[6] 则通过HCK矩阵替代传统的基因表达矩阵,为缺乏参考基因组的复杂群落分析提供了新的思路。
本文侧重介绍smRNA-seq的数据分析工具和框架,从目前通用的scRNA-seq分析流程和工具开始,同时也包括smRNA-seq独有的分析步骤,最后介绍最新的三个分析框架。
smRNA-seq数据分析流程现状
近年来,真核细胞的单细胞RNA测序(scRNA-seq)分析流程已趋于成熟,相关分析工具也呈现爆炸式增长[11]。然而,对于微生物单细胞转录组(smRNA-seq)的研究,大多数仍集中在测序方法上,数据分析部分通常沿用真核细胞的 scRNA-seq工具。如图1所示,本节将以scRNA-seq的通用分析框架为基础,介绍各个分析模块所用的工具,并指出smRNA-seq在方法和工具上的主要差异。
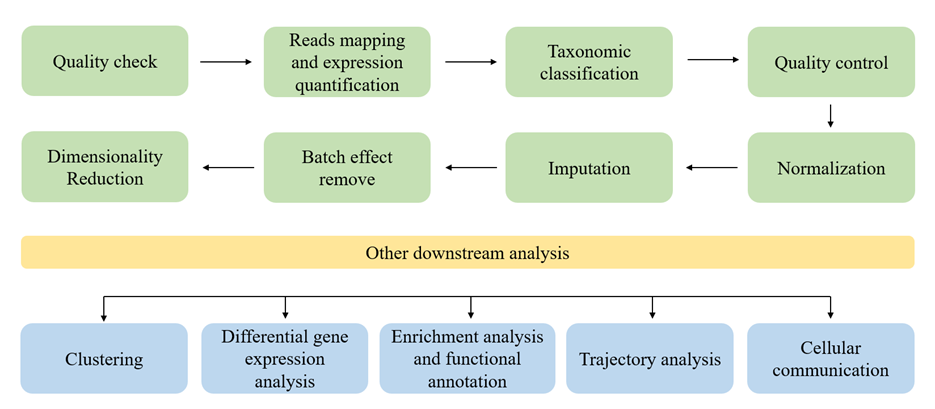
图1 scRNA-seq数据分析流程.
质量检查和参考基因组映射
获取原始测序数据后首先要对数据进行质量控制,为了清洗低质量reads、保留有用信息。目前主要的质量控制工具包括FastQC、Cutadapt和fastp[12–14],FastQC用于检查原始测序数据的总体质量(碱基质量分布、GC含量、接头污染),Cutadapt和fastp属于读长修剪工具。Cutadapt专注于接头去除,适合处理复杂adapter,可以与其他质量控制(Quality Control,QC)工具如FastQC配合使用。Fastp速度快,集成度高,可以做常规QC+trimming一体化处理。基于唯一分子标识符(Unique Molecular Identifier,UMI)的测序,还可以使用UMI-tools[15]利用UMI消除PCR重复,精确基因表达数。
质量控制之后进行读取映射(read mapping)和表达定量以形成基因表达矩阵,需要将每个测序的reads映射到参考基因组并定量表达。读取映射的工具有CellRanger、dropEst、Kallisto-BUStools和Alevin[16–19],它们的核心功能包括读取映射、UMI去重和原始表达计数。其中CellRanger使用率最高,它针对10× Genomics数据优化,具有成熟和完整的流水线,缺点是速度慢且内存要求高。dropEst适用于Drop-seq和inDrop数据,和CellRanger一样支持完整比对。Kallisto-BUStools和Alevin则采用伪比对(pseudoalignment)策略,速度相较于CellRanger显著提升,适合大规模数据处理。对于表达定量的工具,根据捕获的转录本范围,scRNA-seq可分为全长转录本和对于基于3′末端的、基于UMI的scRNA测序。对于生成全长转录本的scRNA-seq,可使用批量RNA-seq软件进行基因或转录本表达定量。基于参考基因组的组装通常比从头组装更准确,常用工具包括Cufflinks、RSEM和StringTie[20–22],其中StringTie在基因/转录本重建和表达定量方面表现最佳。对于基于3′末端的、基于UMI的scRNA测序,常用SAVER[23]。
与真核细胞不同,smRNA-seq通常存在mRNA缺乏poly(A)尾、基因组小、测序深度低以及高噪声背景等特点,质控时需要特别关注rRNA去除效率[3]。在参考基因组映射方面,由于微生物基因组多样性高、可变区多,使用完整、高质量的参考基因组(或非冗余MAG集合)很重要,常需要研究者自己提供参考基因组[6]。
物种注释
与真核单细胞转录组仅需将reads比对到参考基因组不同,微生物单细胞转录组在预处理阶段往往需要额外的物种注释步骤。该环节通常依赖于Kraken2[24] 的k-mer分类框架,并辅以Bracken[25]进行丰度估算。不同流程在数据库选择和注释策略上有所差异:MscT构建基于gOTUs的自定义数据库,并采用“>50% informative reads”的阈值来筛选可信注释;smRandom-seq2的MIC-Anno模块使用UHGG人类肠道基因组数据库[26],并引入root-to-leaf的分层注释策略以提高准确性;mKmer则结合Jellyfish[27]生成高频保守K-mer矩阵,基于标准RefSeq数据库[28] 进行root-to-leaf注释,并通过Bracken和丰度阈值进一步过滤。整体上,微生物单转的物种注释包括制定参考数据库与物种分类策略,是区别于真核单转预处理的核心步骤。
质量控制
scRNA-seq常受到死亡细胞、环境RNA污染及细胞双重捕获等因素的影响,这会导致低质量数据的产生,阻碍下游分析[29],因此应进行第二次质量控制,常用的工具有Seurat、Scanpy和Scater[30–32],它们属于综合性框架,覆盖数据预处理、QC、标准化、降维、聚类等整个scRNA-seq分析流程,也提供一些通用的QC功能(如基因数、UMI、线粒体比例过滤)。还有一些专门化的QC工具,解决特定问题。对于破碎或死亡的低质量细胞,可以使用SinQC[33]和Scater;对于使用液滴测序的方案,常会有环境RNA污染,可以使用SoupX[34] 通过空液滴数据推断污染来源,再结合每个细胞的污染比例,校正基因表达谱,从而去除环境RNA干扰;对于双峰问题,DoubletFinder[35] 通过基因表达特征预测双峰,而Solo[36] 通过半监督深度学习进行双峰检测。最后在进行二次质量控制时,阈值设置需结合生物学背景和组织特性灵活调整。
对于微生物单细胞数据,低质量细胞、环境RNA污染、双峰现象更常见。针对液滴测序,可使用SoupX矫正环境RNA污染,但需要结合物种特异表达模式。与真核不同,smRNA-seq往往需要考虑物种间基因组差异、rRNA比例和基因密度。
归一化
归一化旨在消除测序深度、捕获效率和技术噪声等偏差,保留真实生物学信号。scRNA-seq数据归一化方法会根据是否使用UMI和测序策略有所不同。对于UMI数据,常用方法包括SCTransform(基于负二项回归,同时完成归一化和方差稳定化)和scran(通过计算细胞大小因子校正测序深度差异),这些方法能够有效处理零膨胀和技术噪声,是当前使用最广的工具[37,38]。对于非UMI的全长转录本数据,则常用SC2P(针对每个细胞每个基因的非线性归一化)和SCnorm(基于分位数回归的逆倍积方法),可减少扩增偏差并保留生物学变异[39,40]。相比之下,传统的批量RNA-seq归一化方法如RPKM/FPKM/TPM可以用于全长转录本,但可能引入伪影,不适合单细胞特性。此外,还有一些新方法如Dino、BayNorm和Linnorm[41–43],针对零膨胀、浅层测序和样本异质性等复杂情况提供更鲁棒的归一化解决方案。其中,Dino 通过学习零和非零计数的联合分布来纠正零膨胀;BayNorm 基于贝叶斯推断框架对真实的基因表达进行估计;而 Linnorm 则利用线性模型对技术变异进行建模,以保留生物学异质性。
smRNA-seq零膨胀严重、UMIs覆盖率低,可以使用scran或SCTransform,但需增加为基数或零膨胀模型处理稀疏矩阵。此外微生物细胞体积和mRNA总量差异大,因此计算size factor时要考虑极端值。
插补
scRNA-seq数据中存在大量零值,一部分零值具有生物学意义,另一部分是由于技术问题如逆转录偏差、扩增效率低或测序深度有限导致的非生物零,这些非生物零会影响下游分析,因此需要进行插补以用替代值替换缺失零。针对 scRNA-seq的插补方法包括SAVER、WEDGE、MAGIC、mcImpute、scVI、scGNN、scIGANs和 AutoImpute[44–51]。对于UMI数据,通常被视为非零膨胀,可直接用于降维和聚类,无需插补,但在差异表达分析时可选择mcImpute或MAGIC提升性能;对于非UMI数据适合使用零膨胀模型或插补方法改善降维、聚类和差异表达(Differential Expression,DE)分析效果;而当测序深度足够时,可以省略插补。
smRNA-seq零值多、UMI低,可使用SAVER、MAGIC、scVI等,但需防止过度平滑,保留物种特异表达。这些方法之所以适用于smRNA-seq数据,是因为它们各自具备处理稀疏数据的能力:SAVER使用贝叶斯回归方法估计真实表达值,尤其适用于低丰度计数;MAGIC通过数据扩散(Data Diffusion)在相似细胞间平滑数据,可以有效恢复缺失值;而scVI则利用变分自编码器(VAE)深度学习模型对数据进行概率建模,能够稳健地处理零膨胀和技术噪声。
批次效应矫正
批次效应指在高通量测序或其他实验中,由于实验批次、操作员、设备或时间等非生物学因素引起的数据系统性差异。些差异可能导致同类型细胞在不同批次中显示出不同的基因表达模式,从而掩盖真实的生物学变异,甚至产生假阳性或假阴性的分析结果。目前用于单细胞转录组批次效应校正的工具主要包括Batchelor[52]、Seurat[30]、scVI[48]、BEER、MMD-ResNet和scScope[53–55]。Batchelor和Seurat利用互最近邻(Mutual Nearest Neighbors,MNN)识别不同批次中相似细胞,通过校正向量消除批次偏差;scVI通过变分自动编码器归一化潜在变量,处理批次效应和库大小差异;BEER使用tSNE降维识别批次相关主成分分析(Principal Component Analysis,PCA)子空间并去除,同时保留群体异质性;MMD-ResNet基于残差神经网络最小化不同批次多元分布差异;scScope则通过深度学习的校正层消除批次效应。总体而言,BEER和Batchelor在区分批次效应与生物学差异方面表现优秀,而在复杂细胞类型或大规模数据集情况下,MMD-ResNet、scVI和Seurat可能更具优势。
微生物数据批次效应来源多为培养条件、文库制备、环境差异,深度学习方法在复杂微生物环境样品中更有优势。
降维
一般scRNA-seq降维分析分为两部分,一使用PCA简化数据,二使用t分布随机邻域嵌入(t-distributed Stochastic Neighbor Embedding,t-SNE)或者统一流形近似与投影(Uniform Manifold Approximation and Projection,UMAP)[56,57] 来可视化。此外还有适用于零膨胀数据的零膨胀负二项分布(Zero-Inflated Negative Binomial,ZINB)及扩散映射(Diffusion Map,DM)[58,59]于扩展过程建图,保留连续轨迹信息,更常用于伪时间轨迹分析。PCA属于线性降维,t-SNE、UMAP和DM属于非线性降维技术,可能导致失真和非生物伪影,主要用于可视化。
聚类
scRNA-seq聚类有助于揭示细胞间异质性,识别细胞亚群及其功能状态。聚类分为五种方法:K-means聚类、层次聚类、基于图的聚类、基于密度的聚类和基于深度学习的聚类。使用K-means聚类的工具有SAIC和RaceID[60,61];CIDR、BackSPIN和SINCERA[62–64]是基于分层聚类的聚类工具;基于图的聚类工具有GRACE和CosTaL[65,66];基于密度的聚类工具有GiniClust和Monocle2[67,68];最后DESC、scziDesk、scVAE和scDeepCluster[69]是常用的基于深度学习的聚类工具。每个工具都有各自的优点。SIMLR 和 Monocle计算速度快,SCENIC适合细胞类型鉴定,DESC具有高聚类精度,Secuer性能好,适合大型数据集[70]。因此,工具的选取需要依据实验条件和需求。
smRNA-seq稀疏,K-means或图聚类可能导致小群体丢失,深度学习聚类方法(scDeepCluster、scGNN)更适合捕获微生物亚群和状态异质性。
差异表达分析
差异表达分析用于识别不同细胞群体或条件下显著变化的基因,从而揭示潜在的生物学机制和功能差异。DESeq2和edgeR[71,72] 最初用于批量RNA-seq,目前也广泛适用于scRNA-seq,DESeq2和edgeR都通过广义线性模型(Generalized Linear Model,GLM)分析基因表达差异,DESeq2结合收缩估计稳定方差并使用Wald或似然比检验,而edgeR使用负二项式GLM和条件最大似然估计处理过度离散数据,识别差异表达基因。单细胞RNA-seq差异表达分析面临零膨胀、多模态和缺失值等挑战,为此开发了多种专门工具:如MAST[73] 利用GLM处理双峰分布,Monocle采用Tobit模型和广义加性模型(Generalized Additive Model,GAM)拟合,SCDE基于零膨胀负二项式模型,D3E[74] 通过转录爆发建模,scDD使用多模态贝叶斯建模。其中Monocle灵敏度高但误报多,MAST精度高但灵敏度低,MAST和DESeq2是目前最常用的工具。
零膨胀和测序深度低是微生物单转差异分析的主要问题。一般使用MAST、Monocle,但需调整模型处理稀疏矩阵和小样本群体。
富集分析
富集分析用于系统性的揭示基因或分析特征在生物学功能、信号通路或调控网络中的分布和作用模式。它主要分为三类:过度表达分析(Over-Representation Analysis,ORA)、基因集富集分析(Gene Set Enrichment Analysis,GSEA)、基因集变异分析(Gene Set Variation Analysis,GSVA)。ORA通过统计差异基因在特定功能集中的富集情况来识别显著功能或通路,常用工具包括clusterProfiler、DAVID和g:Profiler[75–77];GSEA不依赖预筛选基因,而是评估功能集在按表达变化排序的全部基因中的集中趋势,以捕捉协调变化信号,常用工具有fgsea、GSEA[78,79] 及clusterProfiler::GSEA[77];GSVA将基因集富集转化为每个样本或细胞的连续评分,用于反映功能活动水平的相对变化,适合单细胞或多样本比较,常用工具为GSVA[80]。
与真核单细胞转录组相比,微生物单细胞转录组在功能注释与富集分析中更依赖于定制化数据库和算法。例如,MscT[83] 流程中基因注释依赖BGMGM,并通过Kraken2/Bracken进行物种分层,功能富集则采用Fisher’s exact test对 KEGG/COG通路进行检验。smRandom-seq2[4]的MIC-Anno流程提出了基于root-to-leaf的分层物种注释方法,并结合UHGG数据库进行注释[26]。smRandom-seq2的MIC-Phage则通过MEME Suite分析标记 K-mer,结合GO富集揭示噬菌体功能。整体上,微生物单细胞研究常用的功能数据库包括KEGG、COG、eggNOG、CAZy、dbCAN等,但因微生物基因功能缺乏完备注释,GSVA等富集方法在单细胞水平应用时需谨慎解释。
轨迹分析
轨迹分析通过重建细胞状态连续变化的路径,揭示细胞发育、分化或疾病进程中的动态过程和关键调控机制。常用的工具有Monocle2[68]、Monocle3[81]和PAGA[82],用于绘制基于基因表达的全局轨迹。近年来,RNA Velocity分析作为一种重要的补充工具被引入,它通过比较未剪接(unspliced)和已剪接(spliced)mRNA的比例,来预测细胞转录状态在短时间内的变化方向和速率,从而提供细胞动态路径的真实方向性信息。
微生物单细胞伪时间分析需先精确筛选特定物种或功能亚群,例如MscT中的B. succiniciproducens细胞。通过Monocle2构建CellDataSet并使用DDRTree降维,结合差异表达基因进行轨迹排序,实现单细胞功能状态的动态变化重建。
细胞通讯分析
细胞通讯分析用于系统性地揭示不同细胞类型之间通过配体-受体相互作用进行信号传递的模式。常用的细胞通讯分析工具包括CellChat、CellPhoneDB、NicheNet、iTALK、LIANA和Connectome[83–88]等。CellChat构建配体-受体网络并支持多条件比较;CellPhoneDB通过统计富集识别显著交互;NicheNet预测信号源对目标细胞基因表达的调控;iTALK筛选差异表达配体-受体对;LIANA整合多数据库方法比较预测结果;Connectome构建网络并分析拓扑结构。其中最常用的是CellChat、CellPhoneDB。
微生物单细胞互作分析依赖于功能注释和物种标注的精确性。例如 MscT 使用SPIEC-EASI构建稀疏逆协方差网络,识别生态关联,并通过Gephi可视化 细胞单位间的交互。
代表性pipeline
smRandom-seq2
smRandom-seq2是2024年由浙江大学王永成团队等建立的基于随机引物的高通量单细胞RNA测序技术及其数据分析方法,首次实现了复杂微生物样本的高通量单细胞转录组测序与分析[4]。
smRandom-seq2的工作流程分为测序和单菌数据分析两部分。测序部分通过基于团队之前开发的smRandom-seq,优化随机引物设计和反应体系,以提高逆转录效率、细菌捕获效率,并有效降低交叉污染。单菌数据分析管道分为三个模块:单微生物注释(MIC-Anno)、细菌分析(MIC-Bac)和噬菌体转录活性分析(MIC-Phage),如图2所示。在MIC-Anno模块中,通过条形码-基因计数散点图确定截止值,利用Kraken2[24] 进行分层注释直至物种水平。MIC-Bac模块中先提取UMI/条码并用STARv2.7.10a、featureCountsv2.0.3[89,90]、UMI-toolsv1.1.2[15] 构建表达矩阵,结合UHGG参考完成比对,再借助Seurat做聚类和差异分析,并用Bowtie2 + GATK进行SNP检测[91,92]。MIC-Phage模块中,以GPD基因组生成gtf,结合SortMeRNAv4.3.4 [93]、STAR、featureCounts、UMI-tools构建矩阵,Seurat v4进行聚类,最终用blastnv2.12.0[94] 预测宿主-噬菌体互作。
总体上,smRandom-seq2在数据分析部分保持了真核单转的分析框架,但更依赖Kraken2、UHGG、blastn等微生物特异工具实现精确注释与功能解析。
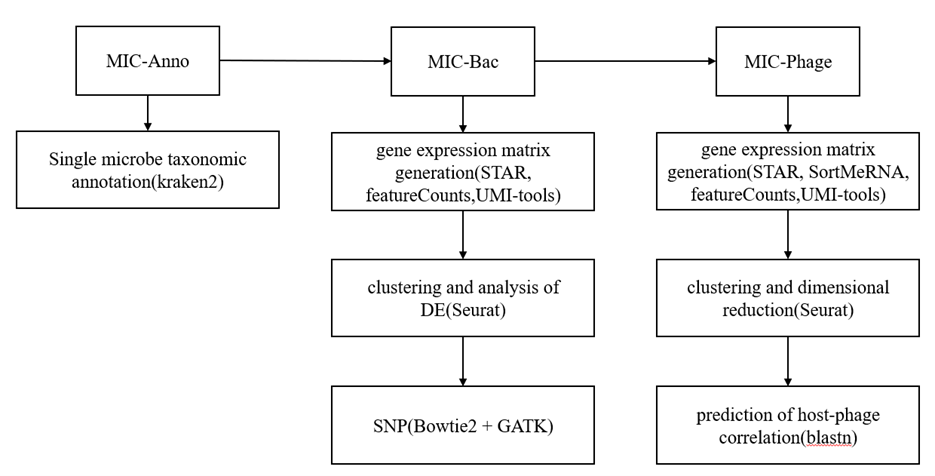
图2 smRandom-seq2分析流程图
MscT
MscT是2024年由浙江大学的孙会增和王永成团队开发的一个微生物单细胞转录组学技术,包括基于液滴的单细胞RNA测序和基于泛基因组的计算分析[5]。首次系统化解决了微生物在单细胞层面缺乏poly(A)尾、基因组参照不足和群体复杂性问题。
该方法的整体流程分为三部分,前端测序,参考基因组数据库构建以及下游分析,本文着重介绍后两部分,如图3所示。
在参考数据库构建上,首先整合55,715个牛胃肠道微生物基因组,用CheckMv1.1.3[95]评估完整度和污染,按MIMAG[96] 标准筛选高、中质量基因组;其次按物种水平聚类生成13,572个非冗余gOTU(dRepv3.2.0,ANI≥95%),分类注释使用GTDB-Tkv2.3.2[97],用PhyloPhlAn3.0+iTOL[98,99] 构建系统发育树;最后,使用Prodigalv2.6.3蛋白质编码基因预测,CD-HIT-EST[100]去冗余,并用COG、KEGG、GO、CAZy进行功能注释,整合得到覆盖牛胃肠微生物的高质量非冗余基因目录。
在预处理部分,首先使用Trimmomaticv0.36[101]进行数据质控与过滤并按条码与UMI分配到单细胞,BWAv0.7.17-r1188/bedtoolsv2.28.0[102,103]用于高质量细胞比对到BGMGM非冗余基因目录,生成单细胞基因表达矩阵。随后进行物种注释,利用Kraken2[24]构建基于gOTU的自定义数据库,并结合Bracken[25]对单细胞reads进行物种注释,从而获得细胞的分类学信息。最后质量控制与高质量细胞筛选通过Seuratv4.3.0完成,同时应用DoubletFinder v2.0.3[35]去除双细胞。下游分析包括聚类、批次效应矫正、功能簇识别、细胞互作、伪时间轨迹和功能富集。高质量基因矩阵导入Seurat进行双细胞过滤、UMAP降维和聚类;Harmony v0.1.0[104]校正批次效应;差异基因由FindAllMarkers[105]识别;SPIEC-EASI[106]分析功能簇互作,特定簇或物种可重新聚类并计算功能基因比例;Monocle2v2.28.0[68]进行伪时间轨迹构建,并基于Fisher’s exact test结合KEGG、COG、GO、CAZy进行功能富集分析。
总体而言MscT的下游分析框架(质控、归一化、降维、聚类、差异分析、功能注释和轨迹推断)与真核单细胞转录组类似,但由于微生物基因组结构与转录特性不同,在工具和参数上有所调整,最大的改变在于采用基于读取数和特征数的质控与 size factor/CLR 归一化,同时下游分析强烈依赖微生物特异的物种分类与功能基因数据库进行注释。
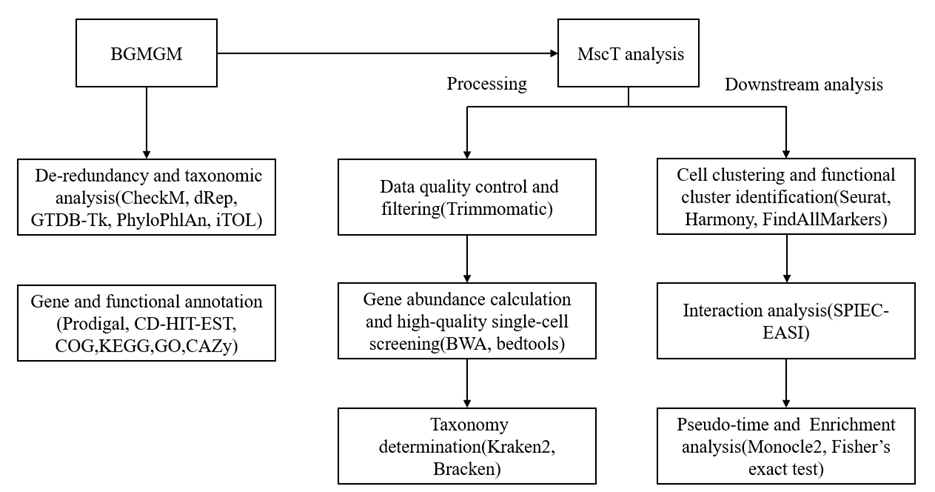
图3 MscT建库及分析流程
mKmer
mKmer是由浙江大学海南研究院樊龙江教授团队在2025年发布的一种基于高频保守K-mer(HCK)的smRNA-seq分析框架[6]。该框架创新的使用HCK矩阵进行细胞分类、物种注释和功能分析,绕过基因或基因组比对,不依赖参考基因组,具体流程如图4所示。
预处理包括质量控制、去冗余、矩阵构建、物种注释、降维、聚类、归一化。首先mKmer使用UMI-tools v1.1.4[15]进行质量控制,对原始测序数据的UMI进行处理。接着使用RemoveDuplicates 去除PCR扩增产生的冗余UMI读段。在矩阵构建部分,利用Jellyfishv2.2.10[27]、KmerFrequency[107]、KmerRank[108]对测序数据进行K-mer计数与分布分析,确定合适的K值并提取高频K-mers,构建了细胞×高频K-mer的频率矩阵;然后使用自研工具KmerCell,结合Jellyfish dump的K-mer计数和清理后的R2 FASTQ数据,选取高频 K-mers并构建了cell-by-HCK矩阵,用于表征每个细胞的K-mer特征。在物种注释部分,使用Kraken2v2.0.7[24]单细胞测序reads进行基于K-mer的逐层分类,并结合 Bracken v2.6.0[25]重新分配read占比,最终通过自研的smAnnotation工具为每个细胞确定物种注释结果。预处理最后使用Seurat对单细胞K-mer表达数据进行归一化、PCA降维、UMAP可视化和SNN聚类,并将Kraken2/Bracken注释结果映射到Seurat对象,同时过滤低丰度或低可信度物种。
下游分析包括功能注释和富集分析。首先使用SortMeRNAv4.2.0[93]去除原始转录组中的rRNA,再用FindAllMarkers[105]提取标记K-mers,并通过MEME套件v5.0.5[109]分析这些高度保守的K-mers基序(K-motifs),最终设计两个工作流程对其进行细菌物种特异性的GO功能富集分析。一个是核苷酸基序分析(KmerGOn)利用MEME套件将K-mers转换为基序文件,匹配已知微生物基序并获取GO功能;另一个蛋白质基序分析(KmerGOp)先用SeqKitv2.8.2[110,111]翻译K-mers为氨基酸序列,再通过MEME、InterProScanv5.47-82.0[112]和AnnotationDbiv1.64.1[113]对蛋白质基序进行GOv3.18.0功能注释,最终系统化揭示每个簇或物种中保守序列的功能。
mKmer使用HCK矩阵代替基因表达矩阵,这是相较于前面smRNA-seq框架的不同点。此外和前两个框架一样,大体上保持了真核单转的分析框架。
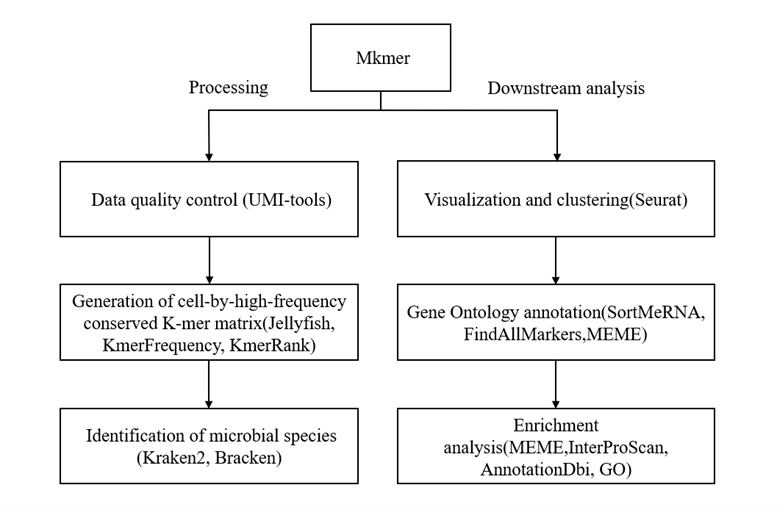
图4 mKmer分析流程图
讨论
在过去的几年里,smRNA-seq技术取得了巨大的进步,虽然在工具上多数沿用scRNA-seq,但是也产生了一些针对微生物的分析框架和思路,很大程度上促进了微生物单细胞转录组学研究,为研究复杂微生物环境中的样本以及微生物与宿主之间的关系提供了更深入的见解。
然而将scRNA-seq技术应用于微生物仍然具有挑战,当前状态下的smRNA-seq数据在稀疏性和测序深度方面与真核生物的scRNA-seq数据存在显著差异。此外,建立良好的基准、最佳实践和和合适的超参数的方法在scRNA-seq中已经被广泛的讨论和确定,对于smRNA-seq尚无类似指南,使得研究者无法立刻得到最合适的参数和方法,影响后续分析。因此,smRNA-seq缺乏一个标准化的分析框架和统一的参数设置,以减轻研究者的分析负担。今后随着更多针对微生物单细胞分析框架的建立和优化,smRNA-seq技术有望得到更广泛的应用。
致谢
该研究由北京林业大学生态与自然保护学院学科交叉融合基金(BH2025-JX-04),中央高校基本科研业务费(XJJSKYQD202546),中国博士后科学基金项目(2024M753580)资助。
利益冲突
作者声明无利益冲突。
参考文献
- Kuchina, A., Brettner, L. M., Paleologu, L., Roco, C. M., Rosenberg, A. B., Carignano, A., Kibler, R., Hirano, M., DePaolo, R. W., Seelig, G., et al. (2021). Microbial single-cell RNA sequencing by split-pool barcoding. Science. 371(6531): eaba5257. https://doi.org/10.1126/science.aba5257
- Blattman, S. B., Jiang, W., Oikonomou, P. and Tavazoie, S. (2020). Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat Microbiol. 5(10): 1192–1201. https://doi.org/10.1038/s41564-020-0729-6
- Xu, Z., Wang, Y., Sheng, K., Rosenthal, R., Liu, N., Hua, X., Zhang, T., Chen, J., Song, M., Lv, Y., et al. (2023). Droplet-based high-throughput single microbe RNA sequencing by smRandom-seq. Nat Commun. 14(1): 5130. https://doi.org/10.1038/s41467-023-40137-9
- Shen, Y., Qian, Q., Ding, L., Qu, W., Zhang, T., Song, M., Huang, Y., Wang, M., Xu, Z., Chen, J., et al. (2024). High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome. Protein Cell. 16(3): 211–226. https://doi.org/10.1093/procel/pwae027
- Jia, M., Zhu, S., Xue, M. Y., Chen, H., Xu, J., Song, M., Tang, Y., Liu, X., Tao, Y., Zhang, T., et al. (2024). Single-cell transcriptomics across 2,534 microbial species reveals functional heterogeneity in the rumen microbiome. Nat Microbiol. 9(7): 1884–1898. https://doi.org/10.1038/s41564-024-01723-9
- Mo, F., Qian, Q., Lu, X., Zheng, D., Cai, W., Yao, J., Chen, H., Huang, Y., Zhang, X., Wu, S., et al. (2025). mKmer: an unbiased K-mer embedding of microbiomic single-microbe RNA sequencing data. Briefings Bioinf. 26(3): e1093/bib/bbaf227. https://doi.org/10.1093/bib/bbaf227
- Imdahl, F., Vafadarnejad, E., Homberger, C., Saliba, A. E. and Vogel, J. (2020). Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat Microbiol. 5(10): 1202–1206. https://doi.org/10.1038/s41564-020-0774-1
- Dar, D., Dar, N., Cai, L. and Newman, D. K. (2021). Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution. Science. 373(6556): eabi4882. https://doi.org/10.1126/science.abi4882
- McNulty, R., Sritharan, D., Pahng, S. H., Meisch, J. P., Liu, S., Brennan, M. A., Saxer, G., Hormoz, S. and Rosenthal, A. Z. (2023). Probe-based bacterial single-cell RNA sequencing predicts toxin regulation. Nat Microbiol. 8(5): 934–945. https://doi.org/10.1038/s41564-023-01348-4
- Ma, P., Amemiya, H. M., He, L. L., Gandhi, S. J., Nicol, R., Bhattacharyya, R. P., Smillie, C. S. and Hung, D. T. (2023). Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell. 186(4): 877–891.e14. https://doi.org/10.1016/j.cell.2023.01.002
- Arya, A., Tripathi, P., Dubey, N., Aier, I. and Kumar Varadwaj, P. (2025). Navigating single-cell RNA-sequencing: protocols, tools, databases, and applications. Genom Inform. 23(1): e1186/s44342–025–00044–5. https://doi.org/10.1186/s44342-025-00044-5
- Andrews, S. (2014). FastQC A Quality Control Tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1): 10. https://doi.org/10.14806/ej.17.1.200
- Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i884–i890. https://doi.org/10.1093/bioinformatics/bty560
- Smith, T., Heger, A. and Sudbery, I. (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27(3): 491–499. https://doi.org/10.1101/gr.209601.116
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun. 8(1): e1038/ncomms14049. https://doi.org/10.1038/ncomms14049
- Petukhov, V., Guo, J., Baryawno, N., Severe, N., Scadden, D. T., Samsonova, M. G. and Kharchenko, P. V. (2018). dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19(1): e1186/s13059–018–1449–6. https://doi.org/10.1186/s13059-018-1449-6
- Melsted, P., Booeshaghi, A. S., Liu, L., Gao, F., Lu, L., Min, K. H., da Veiga Beltrame, E., Hjörleifsson, K. E., Gehring, J., Pachter, L., et al. (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol. 39(7): 813–818. https://doi.org/10.1038/s41587-021-00870-2
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 14(4): 417–419. https://doi.org/10.1038/nmeth.4197
- Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J. and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28(5): 511–515. https://doi.org/10.1038/nbt.1621
- Li, B. and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12(1): e1186/1471–2105–12–323. https://doi.org/10.1186/1471-2105-12-323
- Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T. and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33(3): 290–295. https://doi.org/10.1038/nbt.3122
- Huang, M., Wang, J., Torre, E., Dueck, H., Shaffer, S., Bonasio, R., Murray, J. I., Raj, A., Li, M., Zhang, N. R., et al. (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 15(7): 539–542. https://doi.org/10.1038/s41592-018-0033-z
- Wood, D. E., Lu, J. and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20(1): e1186/s13059–019–1891–0. https://doi.org/10.1186/s13059-019-1891-0
- Lu, J., Breitwieser, F. P., Thielen, P. and Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 3: e104. https://doi.org/10.7717/peerj-cs.104
- Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., Pollard, K. S., Sakharova, E., Parks, D. H., Hugenholtz, P., et al. (2020). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 39(1): 105–114. https://doi.org/10.1038/s41587-020-0603-3
- Marçais, G. and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27(6): 764–770. https://doi.org/10.1093/bioinformatics/btr011
- Pruitt, K. D. (2004). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33: D501–D504. https://doi.org/10.1093/nar/gki025
- Lu, J., Sheng, Y., Qian, W., Pan, M., Zhao, X. and Ge, Q. (2023). scRNA‐seq data analysis method to improve analysis performance. IET Nanobiotechnol. 17(3): 246–256. https://doi.org/10.1049/nbt2.12115
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E. and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 36(5): 411–420. https://doi.org/10.1038/nbt.4096
- Wolf, F. A., Angerer, P. and Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19(1): e1186/s13059–017–1382–0. https://doi.org/10.1186/s13059-017-1382-0
- McCarthy, D. J., Campbell, K. R., Lun, A. T. L. and Wills, Q. F. (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 33(8): 1179–1186. https://doi.org/10.1093/bioinformatics/btw777
- Jiang, P., Thomson, J. A. and Stewart, R. (2016). Quality control of single-cell RNA-seq by SinQC. Bioinformatics. 32(16): 2514–2516. https://doi.org/10.1093/bioinformatics/btw176
- Young, M. D. and Behjati, S. (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 9(12): e1093/gigascience/giaa151. https://doi.org/10.1093/gigascience/giaa151
- McGinnis, C. S., Murrow, L. M. and Gartner, Z. J. (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8(4): 329–337.e4. https://doi.org/10.1016/j.cels.2019.03.003
- Bernstein, N. J., Fong, N. L., Lam, I., Roy, M. A., Hendrickson, D. G. and Kelley, D. R. (2020). Solo: Doublet Identification in Single-Cell RNA-Seq via Semi-Supervised Deep Learning. Cell Syst. 11(1): 95–101.e5. https://doi.org/10.1016/j.cels.2020.05.010
- Hafemeister, C. and Satija, R. (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20(1): e1186/s13059–019–1874–1. https://doi.org/10.1186/s13059-019-1874-1
- Lun, A. T., McCarthy, D. J. and Marioni, J. C. (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 5: 2122. https://doi.org/10.12688/f1000research.9501.2
- Wu, Z., Zhang, Y., Stitzel, M. L. and Wu, H. (2018). Two-phase differential expression analysis for single cell RNA-seq. Bioinformatics. 34(19): 3340–3348. https://doi.org/10.1093/bioinformatics/bty329
- Bacher, R., Chu, L. F., Leng, N., Gasch, A. P., Thomson, J. A., Stewart, R. M., Newton, M. and Kendziorski, C. (2017). SCnorm: robust normalization of single-cell RNA-seq data. Nat Methods. 14(6): 584–586. https://doi.org/10.1038/nmeth.4263
- Brown, J., Ni, Z., Mohanty, C., Bacher, R. and Kendziorski, C. (2021). Normalization by distributional resampling of high throughput single-cell RNA-sequencing data. Bioinformatics. 37(22): 4123–4128. https://doi.org/10.1093/bioinformatics/btab450
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun. 8(1): e1038/ncomms14049. https://doi.org/10.1038/ncomms14049
- Liu, X., Li, L. and Zhao, S. (2019). Linnorm: a robust normalization method for single-cell RNA-seq data. BMC Bioinformatics. 20(1), 312.
- Huang, M., Wang, J., Torre, E., Dueck, H., Shaffer, S., Bonasio, R., Murray, J. I., Raj, A., Li, M., Zhang, N. R., et al. (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 15(7): 539–542. https://doi.org/10.1038/s41592-018-0033-z
- Hu, Y., Li, B., Zhang, W., Liu, N., Cai, P., Chen, F. and Qu, K. (2021). WEDGE: imputation of gene expression values from single-cell RNA-seq datasets using biased matrix decomposition. Briefings Bioinf. 22(5): e1093/bib/bbab085. https://doi.org/10.1093/bib/bbab085
- van Dijk, D., Sharma, R., Nainys, J., Yim, K., Kathail, P., Carr, A. J., Burdziak, C., Moon, K. R., Chaffer, C. L., Pattabiraman, D., et al. (2018). Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 174(3): 716–729.e27. https://doi.org/10.1016/j.cell.2018.05.061
- Mongia, A., Sengupta, D. and Majumdar, A. (2019). McImpute: Matrix Completion Based Imputation for Single Cell RNA-seq Data. Front Genet. 10: e00009. https://doi.org/10.3389/fgene.2019.00009
- Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. and Yosef, N. (2018). Deep generative modeling for single-cell transcriptomics. Nat Methods. 15(12): 1053–1058. https://doi.org/10.1038/s41592-018-0229-2
- Wang, J., Ma, A., Chang, Y., Gong, J., Jiang, Y., Qi, R., Wang, C., Fu, H., Ma, Q., Xu, D., et al. (2021). scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat Commun. 12(1): e1038/s41467–021–22197–x. https://doi.org/10.1038/s41467-021-22197-x
- Xu, Y., Zhang, Z., You, L., Liu, J., Fan, Z. and Zhou, X. (2020). scIGANs: single-cell RNA-seq imputation using generative adversarial networks. Nucleic Acids Res. 48(15): e85–e85. https://doi.org/10.1093/nar/gkaa506
- Talwar, D., Mongia, A., Sengupta, D. and Majumdar, A. (2018). AutoImpute: Autoencoder based imputation of single-cell RNA-seq data. Sci Rep. 8(1): e1038/s41598–018–34688–x. https://doi.org/10.1038/s41598-018-34688-x
- Haghverdi, L., Lun, A. T. L., Morgan, M. D. and Marioni, J. C. (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 36(5): 421–427. https://doi.org/10.1038/nbt.4091
- Zhang, F., Wu, Y. and Tian, W. (2019). A novel approach to remove the batch effect of single-cell data. Cell Discov. 5(1): e1038/s41421–019–0114–x. https://doi.org/10.1038/s41421-019-0114-x
- Shaham, U., Stanton, K. P., Zhao, J., Li, H., Raddassi, K., Montgomery, R. and Kluger, Y. (2017). Removal of batch effects using distribution-matching residual networks. Bioinformatics. 33(16): 2539–2546. https://doi.org/10.1093/bioinformatics/btx196
- Deng, Y., Bao, F., Dai, Q., Wu, L. F. and Altschuler, S. J. (2019). Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat Methods. 16(4): 311–314. https://doi.org/10.1038/s41592-019-0353-7
- Van der Maaten, L. and Hinton, G. (2008). Visualizing data using t-SNE. J Mach Learn Res.9(86):2579−2605. https://www.jmlr.org/papers/v9/vandermaaten08a.html
- McInnes, L., Healy, J., Saul, N. and Großberger, L. (2018). UMAP: Uniform Manifold Approximation and Projection. J open source softw. 3(29): 861. https://doi.org/10.21105/joss.00861
- Risso, D., Perraudeau, F., Gribkova, S. et al. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun 9, 284 (2018). https://doi.org/10.1038/s41467-017-02554-5
- Coifman, R. R. and Lafon, S. (2006). Diffusion maps. Appl Comput Harmon Anal. 21(1): 5–30. https://doi.org/10.1016/j.acha.2006.04.006
- Yang, L., Liu, J., Lu, Q., Riggs, A. D. and Wu, X. (2017). SAIC: an iterative clustering approach for analysis of single cell RNA-seq data. BMC Genomics. 18: e1186/s12864–017–4019–5. https://doi.org/10.1186/s12864-017-4019-5
- Grün, D., Lyubimova, A., Kester, L., Wiebrands, K., Basak, O., Sasaki, N., Clevers, H. and van Oudenaarden, A. (2015). Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 525(7568): 251–255. https://doi.org/10.1038/nature14966
- Zeisel, A., Muñoz-Manchado, A. B., Codeluppi, S., Lönnerberg, P., La Manno, G., Juréus, A., Marques, S., Munguba, H., He, L., Betsholtz, C., et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science (1979). 347(6226): 1138–1142. https://doi.org/10.1126/science.aaa1934
- Lin, P., Troup, M. and Ho, J. W. K. (2017). CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 18(1): e1186/s13059–017–1188–0. https://doi.org/10.1186/s13059-017-1188-0
- Guo, M., Wang, H., Potter, S. S., Whitsett, J. A. and Xu, Y. (2015). SINCERA: A Pipeline for Single-Cell RNA-Seq Profiling Analysis. PLoS Comput Biol. 11(11): e1004575. https://doi.org/10.1371/journal.pcbi.1004575
- Li, Y., Nguyen, J., Anastasiu, D. C. and Arriaga, E. A. (2023). CosTaL: an accurate and scalable graph-based clustering algorithm for high-dimensional single-cell data analysis. Briefings Bioinf. 24(3): e1093/bib/bbad157. https://doi.org/10.1093/bib/bbad157
- Guan, J., Li, R. Y. and Wang, J. (2020). GRACE: A Graph-Based Cluster Ensemble Approach for Single-Cell RNA-Seq Data Clustering. IEEE Access. 8: 166730–166741. https://doi.org/10.1109/access.2020.3022718
- Qiu, X., Hill, A., Packer, J., Lin, D., Ma, Y. A. and Trapnell, C. (2017). Single-cell mRNA quantification and differential analysis with Census. Nat Methods. 14(3): 309–315. https://doi.org/10.1038/nmeth.4150
- Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., Lennon, N. J., Livak, K. J., Mikkelsen, T. S., Rinn, J. L., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 32(4): 381–386. https://doi.org/10.1038/nbt.2859
- Tian, T., Wan, J., Song, Q. and Wei, Z. (2019). Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat Mach Intell. 1(4): 191–198. https://doi.org/10.1038/s42256-019-0037-0
- Wei, N., Nie, Y., Liu, L., Zheng, X. and Wu, H. J. (2022). Secuer: Ultrafast, scalable and accurate clustering of single-cell RNA-seq data. PLoS Comput Biol. 18(12): e1010753. https://doi.org/10.1371/journal.pcbi.1010753
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12): e1186/s13059–014–0550–8. https://doi.org/10.1186/s13059-014-0550-8
- Robinson, M. D., McCarthy, D. J. and Smyth, G. K. (2009). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26(1): 139–140. https://doi.org/10.1093/bioinformatics/btp616
- Finak, G., McDavid, A., Yajima, M., Deng, J., Gersuk, V., Shalek, A. K., Slichter, C. K., Miller, H. W., McElrath, M. J., Prlic, M., et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16(1): e1186/s13059–015–0844–5. https://doi.org/10.1186/s13059-015-0844-5
- Delmans, M. and Hemberg, M. (2016). Discrete distributional differential expression (D3E) - a tool for gene expression analysis of single-cell RNA-seq data. BMC Bioinf. 17(1): e1186/s12859–016–0944–6. https://doi.org/10.1186/s12859-016-0944-6
- Yu, G., Wang, L. G., Han, Y. and He, Q. Y. (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: J Integr Biol. 16(5): 284–287. https://doi.org/10.1089/omi.2011.0118
- Huang, D. W., Sherman, B. T. and Lempicki, R. A. (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4(1): 44–57. https://doi.org/10.1038/nprot.2008.211
- Reimand, J., Kull, M., Peterson, H., Hansen, J. and Vilo, J. (2007). g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35: W193–W200. https://doi.org/10.1093/nar/gkm226
- Korotkevich, G., Sukhov, V., Budin, N., Shpak, B., Artyomov, M. N. and Sergushichev, A. (2016). Fast gene set enrichment analysis. Bioinformatics. 32(14): 2276–2278. https://doi.org/10.1101/060012
- Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 102(43): 15545–15550. https://doi.org/10.1073/pnas.0506580102
- Hänzelmann, S., Castelo, R. and Guinney, J. (2013). GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 14(1): e1186/1471–2105–14–7. https://doi.org/10.1186/1471-2105-14-7
- Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., Zhang, F., Mundlos, S., Christiansen, L., Steemers, F. J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature. 566(7745): 496–502. https://doi.org/10.1038/s41586-019-0969-x
- Wolf, F. A., Hamey, F. K., Plass, M., Solana, J., Dahlin, J. S., Göttgens, B., Rajewsky, N., Simon, L. and Theis, F. J. (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20(1): e1186/s13059–019–1663–x. https://doi.org/10.1186/s13059-019-1663-x
- Jin, S., Plikus, M. V. and Nie, Q. (2024). CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat Protoc. 20(1): 180–219. https://doi.org/10.1038/s41596-024-01045-4
- Efremova, M., Vento-Tormo, M., Teichmann, S. A. and Vento-Tormo, R. (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc. 15(4): 1484–1506. https://doi.org/10.1038/s41596-020-0292-x
- Browaeys, R., Saelens, W. and Saeys, Y. (2019). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 17(2): 159–162. https://doi.org/10.1038/s41592-019-0667-5
- Wang, Y. , Wang, R. , Zhang, S. , Song, S. , & Wang, L. . (2019). Italk: an r package to characterize and illustrate intercellular communication. bioRxiv. https://doi.org/10.1101/507871
- Dimitrov, D., Türei, D., Garrido-Rodriguez, M. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat Commun 13, 3224 (2022). https://doi.org/10.1038/s41467-022-30755-0
- Raredon, M.S.B., Yang, J., Garritano, J. et al. (2022). Computation and visualization of cell–cell signaling topologies in single-cell systems data using Connectome. Sci Rep. 12: 4187 https://doi.org/10.1038/s41598-022-07959-x
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29(1): 15–21. https://doi.org/10.1093/bioinformatics/bts635
- Liao, Y., Smyth, G. K. and Shi, W. (2013). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30(7): 923–930. https://doi.org/10.1093/bioinformatics/btt656
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4): 357–359. https://doi.org/10.1038/nmeth.1923
- McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9): 1297–1303. https://doi.org/10.1101/gr.107524.110
- Kopylova, E., Noé, L. and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 28(24): 3211–3217. https://doi.org/10.1093/bioinformatics/bts611
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol. 215(3):403–410. https://doi.org/10.1016/s0022-2836(05)80360-2
- Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7): 1043–1055. https://doi.org/10.1101/gr.186072.114
- Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., Schulz, F., Jarett, J., Rivers, A. R., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 35(8): 725–731. https://doi.org/10.1038/nbt.3893
- Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 36(6): 1925–1927. https://doi.org/10.1093/bioinformatics/btz848
- Asnicar, F., Thomas, A.M., Beghini, F. et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 11:2500 https://doi.org/10.1038/s41467-020-16366-7
- Letunic, I. and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47: W256–W259. https://doi.org/10.1093/nar/gkz239
- Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W. and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11(1): e1186/1471–2105–11–119. https://doi.org/10.1186/1471-2105-11-119
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15): 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25(14): 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26(6): 841–842. https://doi.org/10.1093/bioinformatics/btq033
- Korsunsky, I., Millard, N., Fan, J., Slowikowski, K., Zhang, F., Wei, K., Baglaenko, Y., Brenner, M., Loh, P. r., Raychaudhuri, S., et al. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 16(12): 1289–1296. https://doi.org/10.1038/s41592-019-0619-0
- Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., Hao, Y., Stoeckius, M., Smibert, P., Satija, R., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell. 177(7): 1888–1902.e21. https://doi.org/10.1016/j.cell.2019.05.031
- Kurtz, Z. D., Müller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., & Bonneau, R. A. (2015).
Sparse and compositionally robust inference of microbial ecological networks.
PLoS Comput Biol. 11(5): e1004226. https://doi.org/10.1371/journal.pcbi.1004226 - Schneider, T. D. and Stephens, R. (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18(20): 6097–6100. https://doi.org/10.1093/nar/18.20.6097
- Chikhi, R. and Rizk, G. (2013). Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol Biol. 8(1): 22. https://doi.org/10.1186/1748-7188-8-22
- Bailey, T. L., Johnson, J., Grant, C. E. and Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43(W1): W39–49. https://doi.org/10.1093/nar/gkv416
- Shen, W., Le, S., Li, Y. and Hu, F. (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One. 11(10): e0163962. https://doi.org/10.1371/journal.pone.0163962
- Shen, W., Sipos, B. and Zhao, L. (2024). SeqKit2: A Swiss army knife for sequence and alignment processing. Imeta. 3(3): e191. https://doi.org/10.1002/imt2.191
- Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., McWilliam, H., Maslen, J., Mitchell, A., Nuka, G., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics. 30(9): 1236–1240. https://doi.org/10.1093/bioinformatics/btu031
- Pagès, H., Carlson, M., Falcon, S. and Li, N. (2025). AnnotationDbi: Manipulation of SQLite-based annotations in Bioconductor. R package version 1.72.0, https://bioconductor.org/packages/AnnotationDbi.
- Kuchina, A., Brettner, L. M., Paleologu, L., Roco, C. M., Rosenberg, A. B., Carignano, A., Kibler, R., Hirano, M., DePaolo, R. W., Seelig, G., et al. (2021). Microbial single-cell RNA sequencing by split-pool barcoding. Science. 371(6531): eaba5257. https://doi.org/10.1126/science.aba5257
- Blattman, S. B., Jiang, W., Oikonomou, P. and Tavazoie, S. (2020). Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat Microbiol. 5(10): 1192–1201. https://doi.org/10.1038/s41564-020-0729-6
- Xu, Z., Wang, Y., Sheng, K., Rosenthal, R., Liu, N., Hua, X., Zhang, T., Chen, J., Song, M., Lv, Y., et al. (2023). Droplet-based high-throughput single microbe RNA sequencing by smRandom-seq. Nat Commun. 14(1): 5130. https://doi.org/10.1038/s41467-023-40137-9
- Shen, Y., Qian, Q., Ding, L., Qu, W., Zhang, T., Song, M., Huang, Y., Wang, M., Xu, Z., Chen, J., et al. (2024). High-throughput single-microbe RNA sequencing reveals adaptive state heterogeneity and host-phage activity associations in human gut microbiome. Protein Cell. 16(3): 211–226. https://doi.org/10.1093/procel/pwae027
- Jia, M., Zhu, S., Xue, M. Y., Chen, H., Xu, J., Song, M., Tang, Y., Liu, X., Tao, Y., Zhang, T., et al. (2024). Single-cell transcriptomics across 2,534 microbial species reveals functional heterogeneity in the rumen microbiome. Nat Microbiol. 9(7): 1884–1898. https://doi.org/10.1038/s41564-024-01723-9
- Mo, F., Qian, Q., Lu, X., Zheng, D., Cai, W., Yao, J., Chen, H., Huang, Y., Zhang, X., Wu, S., et al. (2025). mKmer: an unbiased K-mer embedding of microbiomic single-microbe RNA sequencing data. Briefings Bioinf. 26(3): e1093/bib/bbaf227. https://doi.org/10.1093/bib/bbaf227
- Imdahl, F., Vafadarnejad, E., Homberger, C., Saliba, A. E. and Vogel, J. (2020). Single-cell RNA-sequencing reports growth-condition-specific global transcriptomes of individual bacteria. Nat Microbiol. 5(10): 1202–1206. https://doi.org/10.1038/s41564-020-0774-1
- Dar, D., Dar, N., Cai, L. and Newman, D. K. (2021). Spatial transcriptomics of planktonic and sessile bacterial populations at single-cell resolution. Science. 373(6556): eabi4882. https://doi.org/10.1126/science.abi4882
- McNulty, R., Sritharan, D., Pahng, S. H., Meisch, J. P., Liu, S., Brennan, M. A., Saxer, G., Hormoz, S. and Rosenthal, A. Z. (2023). Probe-based bacterial single-cell RNA sequencing predicts toxin regulation. Nat Microbiol. 8(5): 934–945. https://doi.org/10.1038/s41564-023-01348-4
- Ma, P., Amemiya, H. M., He, L. L., Gandhi, S. J., Nicol, R., Bhattacharyya, R. P., Smillie, C. S. and Hung, D. T. (2023). Bacterial droplet-based single-cell RNA-seq reveals antibiotic-associated heterogeneous cellular states. Cell. 186(4): 877–891.e14. https://doi.org/10.1016/j.cell.2023.01.002
- Arya, A., Tripathi, P., Dubey, N., Aier, I. and Kumar Varadwaj, P. (2025). Navigating single-cell RNA-sequencing: protocols, tools, databases, and applications. Genom Inform. 23(1): e1186/s44342–025–00044–5. https://doi.org/10.1186/s44342-025-00044-5
- Andrews, S. (2014). FastQC A Quality Control Tool for High Throughput Sequence Data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17(1): 10. https://doi.org/10.14806/ej.17.1.200
- Chen, S., Zhou, Y., Chen, Y. and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17): i884–i890. https://doi.org/10.1093/bioinformatics/bty560
- Smith, T., Heger, A. and Sudbery, I. (2017). UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27(3): 491–499. https://doi.org/10.1101/gr.209601.116
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun. 8(1): e1038/ncomms14049. https://doi.org/10.1038/ncomms14049
- Petukhov, V., Guo, J., Baryawno, N., Severe, N., Scadden, D. T., Samsonova, M. G. and Kharchenko, P. V. (2018). dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19(1): e1186/s13059–018–1449–6. https://doi.org/10.1186/s13059-018-1449-6
- Melsted, P., Booeshaghi, A. S., Liu, L., Gao, F., Lu, L., Min, K. H., da Veiga Beltrame, E., Hjörleifsson, K. E., Gehring, J., Pachter, L., et al. (2021). Modular, efficient and constant-memory single-cell RNA-seq preprocessing. Nat Biotechnol. 39(7): 813–818. https://doi.org/10.1038/s41587-021-00870-2
- Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 14(4): 417–419. https://doi.org/10.1038/nmeth.4197
- Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., Salzberg, S. L., Wold, B. J. and Pachter, L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28(5): 511–515. https://doi.org/10.1038/nbt.1621
- Li, B. and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12(1): e1186/1471–2105–12–323. https://doi.org/10.1186/1471-2105-12-323
- Pertea, M., Pertea, G. M., Antonescu, C. M., Chang, T. C., Mendell, J. T. and Salzberg, S. L. (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 33(3): 290–295. https://doi.org/10.1038/nbt.3122
- Huang, M., Wang, J., Torre, E., Dueck, H., Shaffer, S., Bonasio, R., Murray, J. I., Raj, A., Li, M., Zhang, N. R., et al. (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 15(7): 539–542. https://doi.org/10.1038/s41592-018-0033-z
- Wood, D. E., Lu, J. and Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20(1): e1186/s13059–019–1891–0. https://doi.org/10.1186/s13059-019-1891-0
- Lu, J., Breitwieser, F. P., Thielen, P. and Salzberg, S. L. (2017). Bracken: estimating species abundance in metagenomics data. PeerJ Comput Sci. 3: e104. https://doi.org/10.7717/peerj-cs.104
- Almeida, A., Nayfach, S., Boland, M., Strozzi, F., Beracochea, M., Shi, Z. J., Pollard, K. S., Sakharova, E., Parks, D. H., Hugenholtz, P., et al. (2020). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat Biotechnol. 39(1): 105–114. https://doi.org/10.1038/s41587-020-0603-3
- Marçais, G. and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics. 27(6): 764–770. https://doi.org/10.1093/bioinformatics/btr011
- Pruitt, K. D. (2004). NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33: D501–D504. https://doi.org/10.1093/nar/gki025
- Lu, J., Sheng, Y., Qian, W., Pan, M., Zhao, X. and Ge, Q. (2023). scRNA‐seq data analysis method to improve analysis performance. IET Nanobiotechnol. 17(3): 246–256. https://doi.org/10.1049/nbt2.12115
- Butler, A., Hoffman, P., Smibert, P., Papalexi, E. and Satija, R. (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 36(5): 411–420. https://doi.org/10.1038/nbt.4096
- Wolf, F. A., Angerer, P. and Theis, F. J. (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19(1): e1186/s13059–017–1382–0. https://doi.org/10.1186/s13059-017-1382-0
- McCarthy, D. J., Campbell, K. R., Lun, A. T. L. and Wills, Q. F. (2017). Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 33(8): 1179–1186. https://doi.org/10.1093/bioinformatics/btw777
- Jiang, P., Thomson, J. A. and Stewart, R. (2016). Quality control of single-cell RNA-seq by SinQC. Bioinformatics. 32(16): 2514–2516. https://doi.org/10.1093/bioinformatics/btw176
- Young, M. D. and Behjati, S. (2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. GigaScience. 9(12): e1093/gigascience/giaa151. https://doi.org/10.1093/gigascience/giaa151
- McGinnis, C. S., Murrow, L. M. and Gartner, Z. J. (2019). DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 8(4): 329–337.e4. https://doi.org/10.1016/j.cels.2019.03.003
- Bernstein, N. J., Fong, N. L., Lam, I., Roy, M. A., Hendrickson, D. G. and Kelley, D. R. (2020). Solo: Doublet Identification in Single-Cell RNA-Seq via Semi-Supervised Deep Learning. Cell Syst. 11(1): 95–101.e5. https://doi.org/10.1016/j.cels.2020.05.010
- Hafemeister, C. and Satija, R. (2019). Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 20(1): e1186/s13059–019–1874–1. https://doi.org/10.1186/s13059-019-1874-1
- Lun, A. T., McCarthy, D. J. and Marioni, J. C. (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Research. 5: 2122. https://doi.org/10.12688/f1000research.9501.2
- Wu, Z., Zhang, Y., Stitzel, M. L. and Wu, H. (2018). Two-phase differential expression analysis for single cell RNA-seq. Bioinformatics. 34(19): 3340–3348. https://doi.org/10.1093/bioinformatics/bty329
- Bacher, R., Chu, L. F., Leng, N., Gasch, A. P., Thomson, J. A., Stewart, R. M., Newton, M. and Kendziorski, C. (2017). SCnorm: robust normalization of single-cell RNA-seq data. Nat Methods. 14(6): 584–586. https://doi.org/10.1038/nmeth.4263
- Brown, J., Ni, Z., Mohanty, C., Bacher, R. and Kendziorski, C. (2021). Normalization by distributional resampling of high throughput single-cell RNA-sequencing data. Bioinformatics. 37(22): 4123–4128. https://doi.org/10.1093/bioinformatics/btab450
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun. 8(1): e1038/ncomms14049. https://doi.org/10.1038/ncomms14049
- Liu, X., Li, L. and Zhao, S. (2019). Linnorm: a robust normalization method for single-cell RNA-seq data. BMC Bioinformatics. 20(1), 312.
- Huang, M., Wang, J., Torre, E., Dueck, H., Shaffer, S., Bonasio, R., Murray, J. I., Raj, A., Li, M., Zhang, N. R., et al. (2018). SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods. 15(7): 539–542. https://doi.org/10.1038/s41592-018-0033-z
- Hu, Y., Li, B., Zhang, W., Liu, N., Cai, P., Chen, F. and Qu, K. (2021). WEDGE: imputation of gene expression values from single-cell RNA-seq datasets using biased matrix decomposition. Briefings Bioinf. 22(5): e1093/bib/bbab085. https://doi.org/10.1093/bib/bbab085
- van Dijk, D., Sharma, R., Nainys, J., Yim, K., Kathail, P., Carr, A. J., Burdziak, C., Moon, K. R., Chaffer, C. L., Pattabiraman, D., et al. (2018). Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 174(3): 716–729.e27. https://doi.org/10.1016/j.cell.2018.05.061
- Mongia, A., Sengupta, D. and Majumdar, A. (2019). McImpute: Matrix Completion Based Imputation for Single Cell RNA-seq Data. Front Genet. 10: e00009. https://doi.org/10.3389/fgene.2019.00009
- Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. and Yosef, N. (2018). Deep generative modeling for single-cell transcriptomics. Nat Methods. 15(12): 1053–1058. https://doi.org/10.1038/s41592-018-0229-2
- Wang, J., Ma, A., Chang, Y., Gong, J., Jiang, Y., Qi, R., Wang, C., Fu, H., Ma, Q., Xu, D., et al. (2021). scGNN is a novel graph neural network framework for single-cell RNA-Seq analyses. Nat Commun. 12(1): e1038/s41467–021–22197–x. https://doi.org/10.1038/s41467-021-22197-x
- Xu, Y., Zhang, Z., You, L., Liu, J., Fan, Z. and Zhou, X. (2020). scIGANs: single-cell RNA-seq imputation using generative adversarial networks. Nucleic Acids Res. 48(15): e85–e85. https://doi.org/10.1093/nar/gkaa506
- Talwar, D., Mongia, A., Sengupta, D. and Majumdar, A. (2018). AutoImpute: Autoencoder based imputation of single-cell RNA-seq data. Sci Rep. 8(1): e1038/s41598–018–34688–x. https://doi.org/10.1038/s41598-018-34688-x
- Haghverdi, L., Lun, A. T. L., Morgan, M. D. and Marioni, J. C. (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol. 36(5): 421–427. https://doi.org/10.1038/nbt.4091
- Zhang, F., Wu, Y. and Tian, W. (2019). A novel approach to remove the batch effect of single-cell data. Cell Discov. 5(1): e1038/s41421–019–0114–x. https://doi.org/10.1038/s41421-019-0114-x
- Shaham, U., Stanton, K. P., Zhao, J., Li, H., Raddassi, K., Montgomery, R. and Kluger, Y. (2017). Removal of batch effects using distribution-matching residual networks. Bioinformatics. 33(16): 2539–2546. https://doi.org/10.1093/bioinformatics/btx196
- Deng, Y., Bao, F., Dai, Q., Wu, L. F. and Altschuler, S. J. (2019). Scalable analysis of cell-type composition from single-cell transcriptomics using deep recurrent learning. Nat Methods. 16(4): 311–314. https://doi.org/10.1038/s41592-019-0353-7
- Van der Maaten, L. and Hinton, G. (2008). Visualizing data using t-SNE. J Mach Learn Res.9(86):2579−2605. https://www.jmlr.org/papers/v9/vandermaaten08a.html
- McInnes, L., Healy, J., Saul, N. and Großberger, L. (2018). UMAP: Uniform Manifold Approximation and Projection. J open source softw. 3(29): 861. https://doi.org/10.21105/joss.00861
- Risso, D., Perraudeau, F., Gribkova, S. et al. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun 9, 284 (2018). https://doi.org/10.1038/s41467-017-02554-5
- Coifman, R. R. and Lafon, S. (2006). Diffusion maps. Appl Comput Harmon Anal. 21(1): 5–30. https://doi.org/10.1016/j.acha.2006.04.006
- Yang, L., Liu, J., Lu, Q., Riggs, A. D. and Wu, X. (2017). SAIC: an iterative clustering approach for analysis of single cell RNA-seq data. BMC Genomics. 18: e1186/s12864–017–4019–5. https://doi.org/10.1186/s12864-017-4019-5
- Grün, D., Lyubimova, A., Kester, L., Wiebrands, K., Basak, O., Sasaki, N., Clevers, H. and van Oudenaarden, A. (2015). Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 525(7568): 251–255. https://doi.org/10.1038/nature14966
- Zeisel, A., Muñoz-Manchado, A. B., Codeluppi, S., Lönnerberg, P., La Manno, G., Juréus, A., Marques, S., Munguba, H., He, L., Betsholtz, C., et al. (2015). Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science (1979). 347(6226): 1138–1142. https://doi.org/10.1126/science.aaa1934
- Lin, P., Troup, M. and Ho, J. W. K. (2017). CIDR: Ultrafast and accurate clustering through imputation for single-cell RNA-seq data. Genome Biol. 18(1): e1186/s13059–017–1188–0. https://doi.org/10.1186/s13059-017-1188-0
- Guo, M., Wang, H., Potter, S. S., Whitsett, J. A. and Xu, Y. (2015). SINCERA: A Pipeline for Single-Cell RNA-Seq Profiling Analysis. PLoS Comput Biol. 11(11): e1004575. https://doi.org/10.1371/journal.pcbi.1004575
- Li, Y., Nguyen, J., Anastasiu, D. C. and Arriaga, E. A. (2023). CosTaL: an accurate and scalable graph-based clustering algorithm for high-dimensional single-cell data analysis. Briefings Bioinf. 24(3): e1093/bib/bbad157. https://doi.org/10.1093/bib/bbad157
- Guan, J., Li, R. Y. and Wang, J. (2020). GRACE: A Graph-Based Cluster Ensemble Approach for Single-Cell RNA-Seq Data Clustering. IEEE Access. 8: 166730–166741. https://doi.org/10.1109/access.2020.3022718
- Qiu, X., Hill, A., Packer, J., Lin, D., Ma, Y. A. and Trapnell, C. (2017). Single-cell mRNA quantification and differential analysis with Census. Nat Methods. 14(3): 309–315. https://doi.org/10.1038/nmeth.4150
- Trapnell, C., Cacchiarelli, D., Grimsby, J., Pokharel, P., Li, S., Morse, M., Lennon, N. J., Livak, K. J., Mikkelsen, T. S., Rinn, J. L., et al. (2014). The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 32(4): 381–386. https://doi.org/10.1038/nbt.2859
- Tian, T., Wan, J., Song, Q. and Wei, Z. (2019). Clustering single-cell RNA-seq data with a model-based deep learning approach. Nat Mach Intell. 1(4): 191–198. https://doi.org/10.1038/s42256-019-0037-0
- Wei, N., Nie, Y., Liu, L., Zheng, X. and Wu, H. J. (2022). Secuer: Ultrafast, scalable and accurate clustering of single-cell RNA-seq data. PLoS Comput Biol. 18(12): e1010753. https://doi.org/10.1371/journal.pcbi.1010753
- Love, M. I., Huber, W. and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12): e1186/s13059–014–0550–8. https://doi.org/10.1186/s13059-014-0550-8
- Robinson, M. D., McCarthy, D. J. and Smyth, G. K. (2009). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26(1): 139–140. https://doi.org/10.1093/bioinformatics/btp616
- Finak, G., McDavid, A., Yajima, M., Deng, J., Gersuk, V., Shalek, A. K., Slichter, C. K., Miller, H. W., McElrath, M. J., Prlic, M., et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 16(1): e1186/s13059–015–0844–5. https://doi.org/10.1186/s13059-015-0844-5
- Delmans, M. and Hemberg, M. (2016). Discrete distributional differential expression (D3E) - a tool for gene expression analysis of single-cell RNA-seq data. BMC Bioinf. 17(1): e1186/s12859–016–0944–6. https://doi.org/10.1186/s12859-016-0944-6
- Yu, G., Wang, L. G., Han, Y. and He, Q. Y. (2012). clusterProfiler: an R Package for Comparing Biological Themes Among Gene Clusters. OMICS: J Integr Biol. 16(5): 284–287. https://doi.org/10.1089/omi.2011.0118
- Huang, D. W., Sherman, B. T. and Lempicki, R. A. (2008). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4(1): 44–57. https://doi.org/10.1038/nprot.2008.211
- Reimand, J., Kull, M., Peterson, H., Hansen, J. and Vilo, J. (2007). g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35: W193–W200. https://doi.org/10.1093/nar/gkm226
- Korotkevich, G., Sukhov, V., Budin, N., Shpak, B., Artyomov, M. N. and Sergushichev, A. (2016). Fast gene set enrichment analysis. Bioinformatics. 32(14): 2276–2278. https://doi.org/10.1101/060012
- Subramanian, A., Tamayo, P., Mootha, V. K., Mukherjee, S., Ebert, B. L., Gillette, M. A., Paulovich, A., Pomeroy, S. L., Golub, T. R., Lander, E. S., et al. (2005). Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 102(43): 15545–15550. https://doi.org/10.1073/pnas.0506580102
- Hänzelmann, S., Castelo, R. and Guinney, J. (2013). GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC Bioinf. 14(1): e1186/1471–2105–14–7. https://doi.org/10.1186/1471-2105-14-7
- Cao, J., Spielmann, M., Qiu, X., Huang, X., Ibrahim, D. M., Hill, A. J., Zhang, F., Mundlos, S., Christiansen, L., Steemers, F. J., et al. (2019). The single-cell transcriptional landscape of mammalian organogenesis. Nature. 566(7745): 496–502. https://doi.org/10.1038/s41586-019-0969-x
- Wolf, F. A., Hamey, F. K., Plass, M., Solana, J., Dahlin, J. S., Göttgens, B., Rajewsky, N., Simon, L. and Theis, F. J. (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20(1): e1186/s13059–019–1663–x. https://doi.org/10.1186/s13059-019-1663-x
- Jin, S., Plikus, M. V. and Nie, Q. (2024). CellChat for systematic analysis of cell–cell communication from single-cell transcriptomics. Nat Protoc. 20(1): 180–219. https://doi.org/10.1038/s41596-024-01045-4
- Efremova, M., Vento-Tormo, M., Teichmann, S. A. and Vento-Tormo, R. (2020). CellPhoneDB: inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat Protoc. 15(4): 1484–1506. https://doi.org/10.1038/s41596-020-0292-x
- Browaeys, R., Saelens, W. and Saeys, Y. (2019). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 17(2): 159–162. https://doi.org/10.1038/s41592-019-0667-5
- Wang, Y. , Wang, R. , Zhang, S. , Song, S. , & Wang, L. . (2019). Italk: an r package to characterize and illustrate intercellular communication. bioRxiv. https://doi.org/10.1101/507871
- Dimitrov, D., Türei, D., Garrido-Rodriguez, M. et al. Comparison of methods and resources for cell-cell communication inference from single-cell RNA-Seq data. Nat Commun 13, 3224 (2022). https://doi.org/10.1038/s41467-022-30755-0
- Raredon, M.S.B., Yang, J., Garritano, J. et al. (2022). Computation and visualization of cell–cell signaling topologies in single-cell systems data using Connectome. Sci Rep. 12: 4187 https://doi.org/10.1038/s41598-022-07959-x
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M. and Gingeras, T. R. (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29(1): 15–21. https://doi.org/10.1093/bioinformatics/bts635
- Liao, Y., Smyth, G. K. and Shi, W. (2013). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 30(7): 923–930. https://doi.org/10.1093/bioinformatics/btt656
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods. 9(4): 357–359. https://doi.org/10.1038/nmeth.1923
- McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., Garimella, K., Altshuler, D., Gabriel, S., Daly, M., et al. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9): 1297–1303. https://doi.org/10.1101/gr.107524.110
- Kopylova, E., Noé, L. and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 28(24): 3211–3217. https://doi.org/10.1093/bioinformatics/bts611
- Altschul, S. F., Gish, W., Miller, W., Myers, E. W. and Lipman, D. J. (1990). Basic local alignment search tool. J Mol Biol. 215(3):403–410. https://doi.org/10.1016/s0022-2836(05)80360-2
- Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7): 1043–1055. https://doi.org/10.1101/gr.186072.114
- Bowers, R. M., Kyrpides, N. C., Stepanauskas, R., Harmon-Smith, M., Doud, D., Reddy, T. B. K., Schulz, F., Jarett, J., Rivers, A. R., et al. (2017). Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 35(8): 725–731. https://doi.org/10.1038/nbt.3893
- Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. and Parks, D. H. (2019). GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics. 36(6): 1925–1927. https://doi.org/10.1093/bioinformatics/btz848
- Asnicar, F., Thomas, A.M., Beghini, F. et al. (2020). Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat Commun. 11:2500 https://doi.org/10.1038/s41467-020-16366-7
- Letunic, I. and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47: W256–W259. https://doi.org/10.1093/nar/gkz239
- Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W. and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 11(1): e1186/1471–2105–11–119. https://doi.org/10.1186/1471-2105-11-119
- Bolger, A. M., Lohse, M. and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 30(15): 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 25(14): 1754–1760. https://doi.org/10.1093/bioinformatics/btp324
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26(6): 841–842. https://doi.org/10.1093/bioinformatics/btq033
- Korsunsky, I., Millard, N., Fan, J., Slowikowski, K., Zhang, F., Wei, K., Baglaenko, Y., Brenner, M., Loh, P. r., Raychaudhuri, S., et al. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 16(12): 1289–1296. https://doi.org/10.1038/s41592-019-0619-0
- Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., Hao, Y., Stoeckius, M., Smibert, P., Satija, R., et al. (2019). Comprehensive Integration of Single-Cell Data. Cell. 177(7): 1888–1902.e21. https://doi.org/10.1016/j.cell.2019.05.031
- Kurtz, Z. D., Müller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., & Bonneau, R. A. (2015).
Sparse and compositionally robust inference of microbial ecological networks.
PLoS Comput Biol. 11(5): e1004226. https://doi.org/10.1371/journal.pcbi.1004226 - Schneider, T. D. and Stephens, R. (1990). Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18(20): 6097–6100. https://doi.org/10.1093/nar/18.20.6097
- Chikhi, R. and Rizk, G. (2013). Space-efficient and exact de Bruijn graph representation based on a Bloom filter. Algorithms Mol Biol. 8(1): 22. https://doi.org/10.1186/1748-7188-8-22
- Bailey, T. L., Johnson, J., Grant, C. E. and Noble, W. S. (2015). The MEME Suite. Nucleic Acids Res. 43(W1): W39–49. https://doi.org/10.1093/nar/gkv416
- Shen, W., Le, S., Li, Y. and Hu, F. (2016). SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One. 11(10): e0163962. https://doi.org/10.1371/journal.pone.0163962
- Shen, W., Sipos, B. and Zhao, L. (2024). SeqKit2: A Swiss army knife for sequence and alignment processing. Imeta. 3(3): e191. https://doi.org/10.1002/imt2.191
- Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., McWilliam, H., Maslen, J., Mitchell, A., Nuka, G., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics. 30(9): 1236–1240. https://doi.org/10.1093/bioinformatics/btu031
- Pagès, H., Carlson, M., Falcon, S. and Li, N. (2025). AnnotationDbi: Manipulation of SQLite-based annotations in Bioconductor. R package version 1.72.0, https://bioconductor.org/packages/AnnotationDbi.
Please login or register for free to view full text
View full text
Download PDF
Q&A
Copyright: © 2025 The Authors; exclusive licensee Bio-protocol LLC.
引用格式:赵李頔涵, 房楠楠, 王恺毅, 王海燕, 闫利平, 高云云. (2025). 动物微生物单细胞转录组分析工具:现状与未来发展方向.
Bio-101: e1011034. DOI:
10.21769/BioProtoc.1011034.
 How to cite:
How to cite: Zhao, L. D. H., Fang, N. N., Wang, K. Y., Wang, H. Y., Yan, L. P. and Gao, Y. Y. (2025). 动物微生物单细胞转录组分析工具:现状与未来发展方向.
Bio-101: e1011034. DOI:
10.21769/BioProtoc.1011034.
