- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Quantitative Proteomics of Nitrosylated Proteins in Melanoma Using the Biotin-Switch Technique Combined With Tandem Mass Tag Labeling
Published: Vol 15, Iss 23, Dec 5, 2025 DOI: 10.21769/BioProtoc.5535 Views: 1869
Reviewed by: Willy R Carrasquel-UrsulaezRamya VisvanathanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jun 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







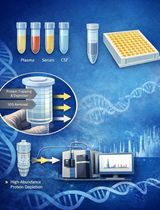
Abstract
Protein S-nitrosylation is a critical post-translational modification that regulates diverse cellular functions and signaling pathways. Although various biochemical methods have been developed to detect S-nitrosylated proteins, many suffer from limited specificity and sensitivity. Here, we describe a robust protocol that combines a modified biotin-switch technique (BST) with streptavidin-based affinity enrichment and quantitative mass spectrometry to detect and profile nitrosylated proteins in cultured cells. The method involves blocking free thiols, selective reduction of nitrosothiols, biotin labeling, enrichment of biotinylated proteins, and identification by tandem mass tag (TMT)-based quantitative mass spectrometry. Additionally, site-directed mutagenesis is employed to generate “non-nitrosylable” mutants for functional validation of specific nitrosylation sites. This protocol provides high specificity, quantitative capability, and versatility for both targeted and global analysis of protein nitrosylation.
Key features
• Specific thiol blocking and labeling: Free thiols are blocked with N-ethylmaleimide, followed by selective reduction and biotinylation of S-nitrosothiols for precise nitrosylation detection.
• Quantitative proteomics: TMT-labeling with high-resolution LC-MS/MS enables multiplexed, accurate quantification and comprehensive nitrosylome profiling with faster data acquisition and fewer missing values than label-free proteomics.
• Functional mutagenesis: Site-directed mutagenesis of cysteine residues generates “non-nitrosylable” mutants to study nitrosylation’s impact on protein function.
• Versatile application: The protocol is adaptable for both targeted protein analysis and global nitrosylation profiling across diverse cell types and experimental conditions.
Keywords: Protein S-nitrosylationGraphical overview

Schematic overview of the biotin switch assay for detection of protein S-nitrosylation. The biotin switch technique (BST) selectively detects S-nitrosylated (SNO) cysteine residues in proteins. Free thiol groups (–SH) are first blocked with N-ethylmaleimide (NEM) to prevent nonspecific labeling. Next, ascorbate selectively reduces S-nitrosothiols (SNO) to free thiols, which are then labeled with biotin-HPDP (N-[6-(biotinamido) hexyl]-3’-(2’-pyridyldithio) propionamide) forming stable disulfide bonds. Biotinylated proteins can be detected either directly by SDS-PAGE (20 μg protein/lane) and immunoblotting using anti-biotin antibodies (1:1,000) and anti-GAPDH (1:3,000, MW: 37 kDa) or enriched by streptavidin bead pulldown for further analysis. In this representative blot, YUGASP cells were pretreated with NOSi (1 mM L-NAME + 200 μM 1400 w acetate) and GSNO (250 μM) for 16 h before lysate preparation and BST procedure.
Background
Snitrosylation, the reversible covalent attachment of nitric oxide (NO) to cysteine thiols (–SH), forming Snitrosothiols (SNO), is a fundamental post-translational modification that regulates protein conformation, activity, localization, and interactions across cellular signaling networks [1]. It influences processes such as mitochondrial quality control, neuroprotection, and vascular tone, and dysregulated Snitrosylation is implicated in diseases including cardiovascular disorders, neurodegeneration, and cancer [2,3]. However, studying Snitrosylation is technically demanding: SNO bonds are labile and exist in low abundance, complicating direct detection. To address this, Jaffrey and Snyder introduced the biotin-switch technique (BST) in 2001. BST employs a three-step workflow: (1) irreversible blocking of free thiols [e.g., with N-ethylmaleimide or MMTS (S-methyl methanethiosulfonate)]; (2) selective reduction of SNO to free thiols using ascorbate; and (3) biotinylation of the newly exposed thiols with biotinHPDP, enabling detection by western blot or affinity enrichment [4,5]. Following its introduction, BST became widely adopted and continues to be the gold standard for both targeted and global Snitrosoproteome analysis. Variants such as SNO-SID, SNORAC, and iodoTMTswitch methods have further enhanced sensitivity and resolution, enabling site-specific identification via mass spectrometry. Complementing these experimental advances, computational tools like GPSSNO 1.0 offer predictive insights for potential Snitrosylation sites. Trained on hundreds of experimentally validated cysteines, GPSSNO achieves approximately 76% accuracy and 54% sensitivity, aiding targeted mutagenesis strategies to confirm functional roles of specific SNO sites [6]. Our current protocol integrates the classic BST with streptavidin-based protein and peptide enrichment, TMT-labeled LC–MS/MS quantitative proteomics, and GPSSNO guided site-directed mutagenesis to generate “nonnitrosylable” protein variants. This combination delivers specificity, quantitative depth, and functional validation, enabling comprehensive investigation of nitrosylation-mediated regulation in diverse cellular systems.
Materials and reagents
Biological materials
1. WM-1366 melanoma cells
2. YUDOSO, YUTICA, and YUGASP patient-derived melanoma cells
Reagents
1. RPMI 1640 medium (Thermo Fisher Scientific, catalog number: 11875-093)
2. FBS (Atlas Biologicals, catalog number: F-0500D)
3. Penicillin-Streptomycin (Thermo Fisher Scientific, catalog number: 15140-122)
4. DPBS (Thermo Fisher Scientific, catalog number: 14190144)
5. Protease inhibitor cocktail (PIC) (Thermo Fisher Scientific, catalog number: 78429)
6. Phosphatase inhibitor cocktail (PHIC) (Thermo Fisher Scientific, catalog number: 78420)
7. HENS buffer, pH ≤ 6.5 (Thermo Fisher Scientific, catalog number: 90106)
8. HEPES solution (Millipore, catalog number: H0887)
9. S-nitroso-L-glutathione (GSNO) (Cayman Chemicals, catalog number: 82240)
10. Diethylenetriamine/DETA-NONOate (Cayman Chemicals, catalog number: 82120)
11. BCA Protein Assay kit (Thermo Fisher Scientific, catalog number: 23227)
12. N-Ethylmaleimide (NEM) (Millipore, catalog number: E1271)
13. Acetone (Sigma, catalog number: 179124)
14. NaCl (Millipore, catalog number: S9888)
15. EDTA (Millipore, catalog number: E4884)
16. L-NAME (Tocris, catalog number: 0665)
17. 1400W dihydrochloride (Selleckchem, catalog number: S8337)
18. L-NIO (Cayman Chemical, catalog number: 80320)
19. L-NIL (Cayman Chemical, catalog number: 80310)
20. CPTIO (Cayman Chemical, catalog number: 81540)
21. Ultrapure water (Millipore, catalog number: DC01L-CS)
22. Biotin-HPDP (Cayman Chemicals, catalog number: 16459)
23. Sodium ascorbate (Sigma, catalog number: PHR1279)
24. Laemmli buffer (Bio-Rad, catalog number: 1610747)
25. Pre-cast 4%–20% SDS-PAGE gels (Bio-Rad, catalog number: 4561091)
26. PVDF membranes (Bio-Rad, catalog number: 162-0184)
27. Non-fat dry milk (Millipore, catalog number: MB9696)
28. 20× TBS buffer (Millipore, catalog number: 20845-M)
29. Tween-20 (Millipore, catalog number: P9416)
30. Triton X-100 (Millipore, catalog number: T8787)
31. HRP-tagged anti-biotin antibodies (Thermo Fisher Scientific, catalog number: 03-3720)
32. Streptavidin magnetic beads (Promega, catalog number: Z548C)
33. Streptavidin agarose resin (Thermo Fisher Scientific, catalog number: 20359)
34. Super Signal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, catalog number: 34080)
35. Total ERK (Cell Signaling Technology, catalog number: 9102S)
36. DUSP4 (Cell Signaling Technology, catalog number: 5149S)
37. RhoC (Cell Signaling Technology, catalog number: 3430S)
38. RSK-1 (Cell Signaling Technology, catalog number: 8408S)
39. GAPDH (Cell Signaling Technology, catalog number: 2118)
40. RASGAP (Proteintech, catalog number: 12935-1-AP)
41. 2-Mercaptoethanol (Millipore, catalog number: M3148)
42. LCMS-grade water (Millipore, catalog number: 1.15333)
43. Dithiothreitol (DTT) (Millipore, catalog number: 3483-12-3)
44. SOLAμTM SPE plates (Thermo Fisher Scientific, catalog number: 60209-001)
45. Trifluoroacetic acid (Millipore, catalog number: 900518)
46. Acetonitrile (ACN) (Millipore, catalog number: 271004)
47. Ammonium bicarbonate (Millipore, catalog number: A6141)
48. Iodoacetic acid (Millipore, catalog number: I4386)
49. Trypsin gold, mass spectrometry grade (Promega, catalog number: V5280)
50. Tandem mass tag (TMT) systems (Thermo Fisher Scientific, catalog number: A40000839)
51. Reversed-phase peptide fractionation cartridge (Thermo Fisher Scientific, catalog number: 84868)
52. Standard human cell line tryptic digest peptide mixture (Promega, catalog number: V6951)
53. Stable isotope-labeled standard peptides (pierce retention time calibration peptide mixture, PRTC) (Thermo Fisher Scientific, catalog number: 88320)
54. Formic acid (Millipore, catalog number: F0507)
Solutions
1. Ascorbate solution (see Recipes)
2. NEM solution (see Recipes)
3. Biotin-HPDP stock (see Recipes)
4. GSNO solution (see Recipes)
5. Neutralization buffer (see Recipes)
6. Wash buffer (see Recipes)
7. Elution buffer (see Recipes)
Recipes
1. Ascorbate solution
Prepare a fresh 50 mM solution of sodium ascorbate in deionized water.
2. NEM solution
Prepare a fresh 400 mM solution of NEM in ethanol.
3. Biotin-HPDP stock
Prepare biotin-HPDP as a 50 mM suspension in DMSO. Freeze at -20 °C until needed.
4. GSNO solution
Prepare a fresh 10 mM GSNO stock solution in PBS, protected from light.
5. Neutralization buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| HEPES (pH 7.7) | 20 mM | 10 mL |
| NaCl | 150 mM | 4.383 g |
| EDTA | 1 mM | 146.12 mg |
| Triton X-100 | 0.5% | 2.5 mL |
| Water (to make up total volume) | n/a | 500 mL |
6. Wash buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| HEPES (pH 7.7) | 20 mM | 10 mL |
| NaCl | 600 mM | 17.532 g |
| EDTA | 1 mM | 146.12 mg |
| Triton X-100 | 0.5% | 2.5 mL |
| Water (to make up total volume) | n/a | 500 mL |
7. Elution buffer
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| HEPES (pH 7.7) | 20 mM | 2 mL |
| NaCl | 100 mM | 584.4 mg |
| EDTA | 1 mM | 29.224 mg |
| β-mercaptoethanol | 5% | 5 mL |
| Water (to make up total volume) | n/a | 100 mL |
8. TBST (Tris-buffered saline and Tween 20)
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Tris | 20 mM | 24.23 g |
| NaCl | 150 mM | 80.06 g |
| Tween 20 | 0.1% | 1 mL |
| Water (to make up total volume) | n/a | 1,000 mL |
Laboratory supplies
1. 100 × 20 mm cell culture dishes (Thermo Fisher Scientific, catalog number: 174903)
2. 25 mL sterile reservoirs (Thermo Fisher Scientific, catalog number: 95128095)
3. Cell scrapers (Thermo Fisher Scientific, catalog number: 179707PK)
4. 15 mL conical tubes (Greiner Bio-One, catalog number: 188261)
5. 50 mL conical tubes (Greiner Bio-One, catalog number: 227261)
6. 5 mL serological pipettes (Greiner Bio-One, catalog number: 606160)
7. 10 mL serological pipettes (Greiner Bio-One, catalog number: 607160)
8. 25 mL serological pipettes (Greiner Bio-One, catalog number: 760160)
9. 1.5 mL microcentrifuge tubes (Greiner Bio-One, catalog number: 616201)
10. Countess cell counting chamber slides (Thermo Fisher Scientific, catalog number: C10283)
Equipment
1. Sonicator (20% amplitude) (Thermo Fisher Scientific, catalog number: FB120110)
2. Heat block (Fisher Scientific, catalog number: 11-686-731)
3. Vortex mixer (Fisher Scientific, catalog number: 02-215-414)
4. UV-visible spectrophotometer (Fisher Scientific, catalog number: NC2027422)
5. BioTek microplate reader (Agilent Technologies, model: Synergy Neo2)
6. Class II biological safety cabinet (Baker, model: SterilGARD® e3)
7. Tissue culture incubator (Fisher Scientific, catalog number: 13-998-124)
8. Inverted microscope (Fisher Scientific, catalog number: LMI3PH4)
9. Tabletop microcentrifuge with rotors for 1.5 mL tubes (Eppendorf, model: 5425/5425 R)
10. Tabletop centrifuge with rotors for 15 mL and 50 mL tubes (Eppendorf, model: 5804/5804 R)
11. Water bath (37 °C) (Fisher Scientific, catalog number: FSSWB27)
12. Countess II FL automated cell counter (Fisher Scientific, model: AMQAF2000)
13. Freezer (-20 °C and -80 °C)
14. SDS-PAGE apparatus (Bio-Rad Mini-PROTEAN Tetra system, catalog number: 1658000)
15. ChemiDoc imaging system (Bio-Rad, model: ChemiDocTM Touch Imaging System)
16. Positive pressure-96 processor (Waters, Milford, MA, catalog number: 186006961)
17. Nanoflow ultra-high-performance liquid chromatograph (RSLCnano, Thermo, San Jose, CA)
18. Electrospray benchtop hybrid quadrupole orbitrap mass spectrometer (Q Exactive HF-X, Thermo, San Jose, CA)
19. SpeedVac, vacuum concentrator (Thermo Scientific, catalog number: 13-875-333)
Software and datasets
1. ImageJ (https://imagej.net/ij/)
2. GraphPad Prism (Dotmatics, https://www.graphpad.com/)
3. Thermo XCalibur or vendor equivalent
4. Skyline for Internal Standard Quantification (https://skyline.ms/project/home/software/Skyline/begin.view) [7].
5. MaxQuant for Database Searching and Quantification (https://maxquant.org/) [8].
6. Mascot Database Searching (Matrix Science)
7. Scaffold (Proteome Software)
Procedure
A. Biotin switch assay for detection of protein S-nitrosylation
Note: This protocol outlines a modified biotin-switch technique for detecting and enriching S-nitrosylated proteins. The method involves selective blocking of free thiols, reduction of S-nitrosothiols, labeling with a biotin derivative, and affinity capture using streptavidin-conjugated beads.
1. Cell harvesting and lysis
a. Wash cells by carefully aspirating the medium and rinsing each culture plate with PBS supplemented with PIC and PHIC to preserve post-translational modifications.
b. Scrape cells using 500 μL of PBS (with PIC and PHIC) per 10 cm dish. Pool cells from designated plates into pre-chilled microcentrifuge tubes.
c. Centrifuge at 1,000× g for 5 min at 4 °C. Discard supernatant.
d. Resuspend the cell pellet in 500 μL of HENS buffer (pH ≤ 6.5), freshly supplemented with PIC and PHIC.
e. Incubate at room temperature for 4 min with gentle inversion to ensure thorough lysis.
f. Sonicate lysate using a probe sonicator with 3 cycles of 3 pulses (1 s on/1 s off) at 20% amplitude.
g. Monitor lysate viscosity and proceed only when lysate is homogeneous, indicating complete DNA shearing and protein solubilization.
2. Clarification of lysates
a. Centrifuge lysates at 15,000× g for 10 min at 4 °C to pellet insoluble debris.
b. Carefully collect the supernatant without disturbing the pellet and transfer to new pre-chilled tubes.
3. Protein quantification and sample storage
a. Reserve 5 μL of each sample for the BCA assay to determine total protein concentration.
b. Store clarified lysates at -20 °C for same-day biotinylation or at -80 °C for longer-term storage. Avoid repeated freeze-thaw cycles.
4. Protein normalization: Normalize all samples to a final protein concentration of 1–1.5 μg/μL, ensuring each contains approximately 1,000 μg of total protein in HENS buffer (pH ≤ 6.5) to maintain consistent biotin-switch efficiency across samples.
5. Positive control treatment: Prepare positive control lysates by treating with GSNO at final concentrations of 10, 50, and 250 μM for 30 min at room temperature. This step verifies the assay’s sensitivity to S-nitrosylation.
6. Blocking free thiols with NEM
a. Prepare a fresh stock of NEM at 50 mg/mL (~400 mM) in ethanol.
b. Add NEM to lysates to a final concentration of 80 mM (e.g., 200 μL of stock per 1 mL of lysate).
c. Incubate at 50 °C for 1 h in the dark, vortexing every 5–10 min to maintain suspension.
d. Replenish NEM halfway through the incubation to ensure complete thiol blocking.
7. Acetone precipitation of proteins
a. Precipitate proteins by adding 4–6 volumes of ice-cold 100% acetone.
b. Invert tubes several times and incubate at -20 °C for 10 min to 1 h.
c. Centrifuge at ~6,000–7,000× g for 10 min at 4 °C.
8. Protein pellet rehydration
a. Air-dry protein pellets at room temperature for 1–2 min (avoid overdrying).
b. Rehydrate pellets first in minimal ultrapure water, then dilute in HENS buffer (with PIC and PHIC, pH ≤ 6.5) to the original sample volume.
9. Biotin labeling of S-nitrosylated cysteines
a. Add sodium ascorbate to a final concentration of 40 mM to selectively reduce S-nitrosothiols to free thiols (e.g., add 40 μL from a 1 M stock to a 1 mL sample).
b. Immediately add Biotin-HPDP to a final concentration of 1 mM (e.g., 20 μL from a 50 mM stock per 1 mL of sample).
c. Incubate at room temperature for 1–2 h in the dark with gentle mixing.
10. Removal of excess reagents via second acetone precipitation
a. Repeat steps A7–8 to remove unreacted biotin and salts.
b. Resuspend protein pellets in HENS buffer (pH ≤ 6.5) to a consistent volume for downstream enrichment.
11. Streptavidin affinity enrichment
a. Prepare streptavidin agarose beads by washing 2–3 times with neutralization buffer.
b. Add 2 volumes of neutralization buffer to each protein sample.
c. Add beads at a ratio of 20–50 μL of beads per milligram of protein input.
d. Incubate samples for 1 h at room temperature or overnight at 4 °C with gentle rotation.
12. Bead washing
a. Wash beads five times with 5 volumes of wash buffer. After each wash:
i. Centrifuge at 200× g for 5 min at room temperature.
ii. Carefully remove supernatant without disturbing the beads.
13. Elution of biotinylated proteins
a. Elute enriched S-nitrosylated proteins using either method:
i. Mild elution: Incubate beads with elution buffer containing 100 μM β-mercaptoethanol to selectively cleave the disulfide bond between Biotin-HPDP and cysteine.
ii. Denaturing elution: Add 1× Laemmli buffer with 5% β-mercaptoethanol, boil for 10 min at 95 °C, and centrifuge to collect supernatant.
14. SDS-PAGE and immunoblotting for biotinylated proteins (Figures 1–2)
a. Use 20 μg of enriched eluted protein sample.
b. Separate proteins by SDS-PAGE on an appropriate polyacrylamide gel.
c. Transfer to a PVDF membrane using wet or semi-dry transfer apparatus.
d. Block membrane in 5% non-fat dry milk (NFDM) in TBST for 1 h at room temperature.
e. Incubate overnight at 4 °C with primary antibody diluted 1:1,000 in 5% NFDM dissolved in 1× TBST.
f. Wash membrane five times for 5 min each with 1× TBST.
g. Incubate with HRP-conjugated secondary antibody (1:5,000 in 5% NFDM dissolved in 1× TBST) for 1 h at room temperature.
h. Wash as above to remove unbound secondary antibody.
i. Incubate with chemiluminescent HRP substrate as per the manufacturer’s instructions.
j. Visualize using a chemiluminescence imager.

Figure 1. Detection of the nitrosylated proteome in YUGASP cells using a biotin-switch technique (BST). Whole cell lysates from YUGASP cells were subjected to the BST assay to identify protein S-nitrosylation under different treatment conditions. (A) Coomassie blue staining confirms equal protein loading across lanes, (B) Immunoblot of biotinylated proteins shows the global pattern of nitrosylated proteins under control (Ctrl), trametinib (Tr), NOS inhibitor (NOSi), NOSi + Tr, LNIO + LNIL, LNIO + LNIL + Tr, and CPTIO conditions. The protein ladder is indicated in kDa on the left. (C) Relative intensity of the nitrosylated proteome from (B). Each sample was normalized to its corresponding Coomassie Blue loading.

Figure 2. Biotin switch assay for the detection of protein S-nitrosylation. (A) S-nitrosylation was induced in WM1366 cell lysates by treatment with increasing concentrations of S-nitrosoglutathione (GSNO; 0, 10, 50, and 250 μM) for 4 h in a dose-dependent manner. The S-nitrosylated proteome was subsequently labeled with biotin via the biotin-switch technique. Biotin-tagged S-nitrosylated proteins were enriched using streptavidin-conjugated agarose beads and analyzed by immunoblotting for target proteins of interest. Total GAPDH and total ERK served as loading controls for input lysates. (B) Densitometric analyses of >3 biological repeats of western blots shown in (A).
15. Data analysis
a. Visualize images using a Bio-Rad chemiluminescence imager (Figures 1–2).
b. Quantify relative band intensities using ImageJ.
c. Normalize target protein signals to input controls and then to untreated control samples.
d. Plot data using GraphPad Prism.
16. Site-directed mutagenesis of targeted cysteine residues: As we followed in [9], potential nitrosylation sites can be predicted using GPS-SNO 1.0 software. Selected cysteine residues were mutated to alanine by site-directed mutagenesis. Wild-type and mutant constructs were transfected into HEK293 cells with Lipofectamine 3000. Nitrosylation status was evaluated by the biotin switch technique (BST), followed by affinity purification and immunoblotting. Functional consequences of the mutations on phosphorylation and downstream signaling were assessed in the presence or absence of nitric oxide donors [9].
B. Mass spectrometry for the detection of protein S-nitrosylation
1. Proteins containing S-nitrosylated (SNO) cysteine residues are selectively labeled using the biotin-switch technique as described above.
2. Add 50 μL of 50% high-capacity streptavidin agarose resin slurry (ThermoFisher 20357) to each protein sample and incubate for 1 h at room temperature with constant mixing on a rotator. This step captures the biotinylated proteins.
3. Centrifuge samples at 1,000× g for 1 min to pellet resin and discard supernatant.
4. Wash resin four times with 500 μL of PBS.
5. Wash resin four times with 500 μL of LC–MS-grade water.
6. Elute proteins by adding 100 μL of elution buffer (aqueous 0.1% TFA, 50% ACN) and incubating the samples for 3 min. Transfer the eluate to a new microcentrifuge tube. Repeat this step two additional times, combining all elutions.
7. Dry the eluted proteins with vacuum centrifugation without heating using the SpeedVac.
8. Re-dissolve the samples in aqueous 50 mM ammonium bicarbonate pH 7.4.
9. Add DTT to reduce disulfide bonds (to a final concentration of 2 mM) and incubate at 60 °C for 20 min.
10. Cool samples to room temperature and add iodoacetic acid to alkylate free cysteines (to 20 mM final concentration). Incubate for 15 min in the dark.
11. Add trypsin (in a 1:20 ratio to total protein) to the reduced and alkylated samples and digest overnight at 37 °C.
12. Acidify the tryptic peptides with aqueous 1% TFA and desalt with SOLA plates following the manufacturer's instructions using a Waters Positive pressure-96 processor.
13. TMT label (TMT 10plex) each sample, minimizing crosstalk by placing similar samples in the 13C channels and another set of similar samples in the 15N channels (e.g., control and experimental conditions). Check labeling with LC–MS/MS using 5% of each sample to ensure at least 95% label incorporation using spectral counting of unmodified and TMT-labeled peptides as a surrogate for quantification of unmodified vs. TMT-modified peptide ion signals (Table 1).
Note: TMT reagents have lot-to-lot variation in the crosstalk between channels, which can be determined from the vendor’s website (https://www.thermofisher.com/order/catalog/product/90110). In this example, three melanoma cell lines are used to provide replicates with distinct biological backgrounds rather than using 3 replicates of control and treatment of the same cell line.
Table 1. Example tandem mass tag layout and labeling quality control . The experimental design included three cell lines with DMSO vehicle control or GSNO treatment; an inhibitor treatment and a negative control were also included. The percentage of labeling was estimated using spectral counting to determine the percentage of TMT-derivatized peptides to the total number of detected peptides. Here, three patient-derived NRAS-mutant melanoma cell lines are used rather than three replicates of a single cell line.
| Sample | Condition | Mass tag | Reporter ion | Labeling (%) |
|---|---|---|---|---|
| YUDOSO | Vehicle control | TMT10-126 | 126.127726 | 94.8 |
| YUDOSO | GSNO | TMT10-127N | 127.124761 | 99.8 |
| YUTICA | Vehicle control | TMT10-127C | 127.131081 | 98.9 |
| YUTICA | GSNO | TMT10-128N | 128.128116 | 100 |
| YUGASP | Vehicle control | TMT10-128C | 128.134436 | 99.6 |
| YUGASP | GSNO | TMT10-129N | 129.131471 | 99.8 |
| Blank | TMT10-129C | 129.13779 | N/A | |
| Blank | TMT10-130N | 130.134825 | N/A | |
| Inhibitor | TMT10-130C | 130.141145 | 98.1 | |
| Negative control | TMT10-131 | 131.13818 | 99.9 |
15. Pool samples using equal amounts of input and dry by vacuum centrifugation without heating.
16. Re-dissolve the TMT-labeled peptide mixture in PBS.
17. Add 50 μL of 50% high-capacity streptavidin agarose resin slurry to each peptide sample and incubate for 1 h at room temperature with constant mixing on a rotator. This step captures the biotinylated peptides.
18. Centrifuge samples at 1,000× g for 1 min to pellet resin and keep the supernatant (unmodified peptides from nitrosylated proteins that were enriched with the biotin switch technique).
19. Wash the resin four times with 500 μL of PBS.
20. Wash the resin four times with 500 μL of LC–MS-grade water.
21. Elute proteins by adding 100 μL of elution buffer (aqueous 0.1% TFA, 50% ACN) and incubating the sample for 3 min at room temperature. Transfer the elute to a new microcentrifuge. Repeat this step two additional times, combining the elutions.
22. Dry the eluate by vacuum centrifugation (SpeedVac) without heating and re-dissolve in 2% ACN + 0.1% formic acid for mass-spec analysis.
23. bRPLC fractionation of unmodified peptides was performed using a high-pH reverse-phase peptide fractionation cartridge according to the manufacturer’s instructions.
24. Perform LC–MS/MS analysis for both the derivatized peptides (1 sample from the second peptide-level pulldown) and for the unmodified peptides (4 fractions from the first protein-level pulldown) to get information about the amino acids that have undergone the biotin switch for cysteine nitrosylation and their proteins of origin.
Note: In our work, a nanoflow ultra-high-performance liquid chromatograph (RSLCnano, Thermo, San Jose, CA) coupled to an electrospray benchtop orbitrap mass spectrometer (Q Exactive HF-X, Thermo, San Jose, CA) was used for tandem mass spectrometry peptide sequencing experiments. The sample was first loaded onto a pre-column (C18 PepMap100, 2 cm × 100 μM ID, 5 μM particle size, 100 Å pore size) and washed for 8 min with aqueous 2% acetonitrile and 0.1% formic acid. The trapped peptides were eluted onto the analytical column (C18 PepMap100, 75 μM ID × 25 cm, 2 μM particle size, 100 Å pore size, Thermo). The 120-min gradient was programmed as 95% solvent A (aqueous 2% acetonitrile + 0.1% formic acid) for 8 min, solvent B (aqueous 90% acetonitrile + 0.1% formic acid) from 5% to 38.5% in 90 min, then solvent B from 50% to 90% in 7 min and held at 90% for 5 min, followed by solvent B from 90% to 5% in 1 min and re-equilibration for 10 min. The flow rate on the analytical column was 300 nL/min. Spray voltage was 1,900 V. Capillary temperature was 275 °C. S-lens RF level was set at 40. The top 20 tandem mass spectra were collected in a data-dependent manner following each survey scan. The resolution values for MS and MS/MS scans were set at 60,000 and 45,000, respectively; resolution for MS/MS was determined by the need to separate the 13C- and 15N-labeled peaks of the TMT reporter ions. Dynamic exclusion was 15 s for previously sampled peptide peaks.
25. Review LC–MS/MS to evaluate instrument performance using vendor software (XCalibur/Freestyle, Thermo). Quantify internal standard peptides (PRTC) using Skyline software or equivalent to show equivalent detection across samples and fractions (Figure 3).
26. Data analysis: Perform database searches for all LC–MS/MS proteomics data.
Note: In our work, MaxQuant (version 1.6.2.10) was used to identify peptides and quantify the TMT reporter ion intensities. The protein database was downloaded from UniProt in March 2021. Up to two missed trypsin cleavages were allowed. Carbamidomethyl cysteine and methionine oxidation were set as variable modifications; the modification of the biotin switch technique, biotin-HPDP-cysteine, and TMT modifications of lysines and peptide N-termini were also added as variable modifications to the database search. Both peptide spectral match (PSM) and protein false discovery rate (FDR) were set at 0.01. The match between runs feature was activated to use MS/MS data from each run to identify peaks by their retention time and mass-to-charge ratios when MS/MS data were not available. Example datasets are available through PRIDE (https://www.ebi.ac.uk/pride/archive/projects/PXD050244); data uploads were searched with Mascot (Matrix Science) using the parameters described above.
27. Data Evaluation: Review the MaxQuant protein file to evaluate protein-level information from the pulldown (Figure 3).
Note: In this experiment, 3,747 proteins were detected. Removal of reverse sequence hits, which are used to establish the false discovery rate, and contaminant proteins (e.g., from culture media or keratins) left 3,681 proteins; further filtering for detection of two or more peptides filtered the list to 2,995 proteins. For the HPDP-biotinylation, 114 modified peptides were detected with one reverse sequence (FDR 1.75%, comparable to the 1% FDR set for peptide spectral match and protein in the database search parameters). Examination of these two datasets showed that 2,906 proteins only had evidence at the protein level, 89 proteins had HPDP-biotinylated peptides and additional unmodified peptides, and 5 proteins were only detected by their HPDP-biotinylated peptides.
28. Log2-transform the data for further analysis.
Note: GSNO increased the levels of proteins recovered using the BST for each cell line. Average log2 ratios for GSNO compared to treatment control were 4.33 for YUDOSO, 3.17 for YUTICA, and 5.8 for YUGASP. Evaluation of this metric indicates that the treatment is having the desired effect in increasing nitrosylation and subsequent biotin-switching. Additional filtering using log2 ratio values could increase the confidence in the assignments for nitrosylated proteins in this dataset, as would the selection of the proteins and peptides that are consistently increased after GSNO treatment in all three cell lines. For modified peptides, manual inspection of the tandem mass spectra is critical for confirmation of sequence assignments and modifications prior to biological follow-up experiments using site-directed mutagenesis or pharmacological inhibition to test the relevance of specific nitrosylation sites or the function and downstream impact of protein nitrosylation.

Figure 3. Example mass spectrometry data for biotinylated peptide identification. (A) The mass spectrum shows a triply charged peptide observed at m/z 762.0859. (B) The tandem mass spectrum identifies the peptide as AAVEEGIVLGGGC442ALLR from mitochondrial 60 kDa heat shock protein (CH60_HUMAN), where the N-terminus is labeled with TMT, and the cysteine is modified by HPDP-biotylilation. The precursor mass measurement accuracy is -1.9 ppm. The peptide spectral match was made by MaxQuant software with a score of 72.2 in the Andromeda search algorithm. (C) The relative quantification for the peptide is also derived from the tandem mass spectrum.
General notes and troubleshooting
General notes
1. Acetone precipitation leads to protein sample loss, which requires complete pellet resuspension and minimizing drying of the pellet.
2. Bead handling: Do not let beads dry during washes. The use of low-speed centrifugation at 200× g helps prevent excessive loss during the process.
3. Specificity of S-nitrosylation detection depends on including both positive (GSNO-treated) and negative controls (no ascorbate) in every experiment.
4. NEM shows thiol specificity at pH below 7.5, but it reacts with amines at higher pH values, so proper adjustments should be made.
5. The biotin-HPDP molecule can break down into its components: β-mercaptoethanol releases biotinylated proteins, which prevents their detection by streptavidin-based methods. The anti-biotin antibody needs to be validated for immunoblotting applications.
Troubleshooting
A. BST method
1. Low or no signal.
a. The use of old or deteriorated reagents should be avoided because it requires fresh sodium ascorbate and biotin-HPDP.
b. The blocking process may be incomplete because NEM needs to be fresh during the incubation period, and the concentration should be doubled.
c. SNOs degrade when samples are left at room temperature under light exposure; so, maintain pH levels at 6.5 or lower and perform all procedures on ice.
d. The labeling process requires longer incubation periods (up to 2 h) while checking that all reagents remain in solution.
e. The loss of proteins can be prevented by being careful with the collection of pellets after the spin process and by minimizing the number of bead washing steps.
2. High background/nonspecific signal.
a. The presence of unblocked thiols needs to be checked through both NEM concentration measurement and verification of proper incubation times.
b. The process of biotinylation shows nonspecific binding when ascorbate is used as a control to establish the SNO-dependent process.
c. The selection of blocking buffer must be appropriate, while controls should be included to avoid antibody cross-reactivity.
3. Poor protein recovery.
a. The process of air drying should be limited to a brief period of 1–2 min followed by immediate rehydration of the pellets.
b. Rehydration becomes complete when the solution reaches 37 °C and the pellet gets dissolved by vortexing or pipetting.
4. Weak or inconsistent western blot.
a. The unequal distribution of proteins in samples can be fixed by BCA normalization and Ponceau S staining of the membrane.
b. The reaction fails when antibodies or substrate become inactive, so fresh dilutions must be prepared while avoiding multiple freeze-thaw cycles.
c. The imaging process requires both exposure time adjustments as well as verification of positive control results.
5. Smearing or extra bands.
a. The combination of ice storage and PIC/PHIC addition in all buffers protects against proteolysis.
b. The sample must contain no more than 20 µg of protein per lane while being thoroughly mixed with sample buffer.
c. The sample needs another acetone precipitation to remove salt contaminants.
6. Low pulldown efficiency.
a. The use of 20–50 μL of beads per milligram of protein with pre-washed beads produces optimal results for this process.
b. The use of control lysates should be combined with input/IP verification to determine if endogenous biotinylated proteins are present.
B. Liquid chromatography–tandem mass spectrometry
1. Instrument performance
a. Evaluate LC–MS/MS performance with a standard sample that will evaluate peptide sequencing capability. Digests of K562 lysate can be used to determine whether consistent numbers of peptides and proteins are identified in each LC–MS/MS analysis. Poor performance can be corrected by evaluating the stability of electrospray ionization, cleaning the mass spectrometer, or evaluating additional quality metrics for ion transmission.
b. Add internal standards, like the peptide retention time calibrator mix, to each sample to examine whether the ion signal is stable across the batch of samples. Ion signal for these peptides can be monitored with different academic and vendor software applications (Skyline). Correct if needed as described in the previous step.
c. Evaluate LC separation by examining peak widths and peak shapes. If needed, replace the solvents or columns to improve separation.
d. If no signal is observed from the standards or sample, evaluate sample pickup in the autosampler using known amounts of liquid in each vial to quantify the sample amount injected.
e. Address low-quality MS/MS data by evaluating the overall ion signal as above and using standard samples to indicate insufficient instrument performance. Evaluate different collision energies when switching from label-free proteomics to TMT proteomics.
f. If large amounts of streptavidin peptides are observed in the LC–MS/MS data, change to monomeric avidin beads (e.g., Pierce NeutrAvidin Agarose Beads, Thermo) or magnetic beads to limit avidin leaching upon protein elution.
g. Examine the crosstalk between TMT channels to minimize “bleed over” from one sample to another. In this example, we used the TMT channels with 13C and 15N isotopes for vehicle controls and GSNO treatments, respectively, to avoid contamination of treated samples in the controls.
h. To expand to large sample numbers, optimize sample handling in 96-well plates, and design a control sample (often a pool of available samples) for normalization of batch effects. TMT reagents have expanded from 10-plex to 16-, 18-, 32-, and 35-plex. Note that 32- and 35-plex TMT reagents use deuterium labeling and require additional controls to compare samples with hydrogen-labeled TMT tags and deuterium-labeled TMT tags due to the slight differences in LC elution profiles.
Acknowledgments
Author contributions:
V.K.Y.: Performed the experiments and data analyses and drafted the initial version of the biotin-switch technique (BST) procedure. B.F. and J.K.: Conducted mass spectrometry experiments for nitrosylated proteins and prepared the corresponding methods section. S.P. and J.S.: Conceived and designed the overall study and experiments, provided supervision, performed formal analyses, and contributed to manuscript writing, review, and editing. All authors reviewed and approved the final manuscript.
Funding was provided by the NIH-R21 grant (R21ES035196) and Moffitt Cancer Center Support account (for S. Premi). The Proteomics & Metabolomics Core is supported in part by the NCI Cancer Center Support Grant (P30-CA076292), which confers Moffitt’s status as a Comprehensive Cancer Center.
The graphical overview was created in BioRender (https://BioRender.com/pe77sdz).
Competing interests
There is no conflict of interest.
References
- Srivastava, J., Yadav, V. K., Jimenez, R. V., Phadatare, P. R., Inamdar, N. A., Young, M. M., Bacchiocchi, A., Halaban, R., Fang, B., Pulido, A. d. M., et al. (2025). Blocking Nitrosylation Induces Immunogenic Cell Death by Sensitizing NRAS-Mutant Melanoma to MEK Inhibitors. Cancer Res. 85(12): 2268–2287. https://doi.org/10.1158/0008-5472.can-24-0693
- Stomberski, C. T., Hess, D. T. and Stamler, J. S. (2019). Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid Redox Signal. 30(10): 1331–1351. https://doi.org/10.1089/ars.2017.7403
- Foster, M. W., Hess, D. T. and Stamler, J. S. (2009). Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 15(9): 391–404. https://doi.org/10.1016/j.molmed.2009.06.007
- Jaffrey, S. R. and Snyder, S. H. (2001). The Biotin Switch Method for the Detection of S-Nitrosylated Proteins. Sci Signaling. 2001(86): pl1–pl1. https://doi.org/10.1126/scisignal.862001pl1
- Forrester, M. T., Foster, M. W., Benhar, M. and Stamler, J. S. (2009). Detection of protein S-nitrosylation with the biotin-switch technique. Free Radical Biol Med. 46(2): 119–126. https://doi.org/10.1016/j.freeradbiomed.2008.09.034
- Xue, Y., Liu, Z., Gao, X., Jin, C., Wen, L., Yao, X. and Ren, J. (2010). GPS-SNO: Computational Prediction of Protein S-Nitrosylation Sites with a Modified GPS Algorithm. PLoS One. 5(6): e11290. https://doi.org/10.1371/journal.pone.0011290
- MacLean, B., Tomazela, D. M., Shulman, N., Chambers, M., Finney, G. L., Frewen, B., Kern, R., Tabb, D. L., Liebler, D. C., MacCoss, M. J., et al. (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics. 26(7): 966–968. https://doi.org/10.1093/bioinformatics/btq054
- Cox, J. and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26(12): 1367–1372. https://doi.org/10.1038/nbt.1511
- Srivastava, J., Yadav, V. K., Jimenez, R. V., Phadatare, P. R., Inamdar, N. A., Young, M. M., Bacchiocchi, A., Halaban, R., Fang, B., Pulido, A. d. M., et al. (2025). Blocking Nitrosylation Induces Immunogenic Cell Death by Sensitizing NRAS-Mutant Melanoma to MEK Inhibitors. Cancer Res. 85(12): 2268–2287. https://doi.org/10.1158/0008-5472.can-24-0693
Article Information
Publication history
Received: Aug 4, 2025
Accepted: Oct 19, 2025
Available online: Dec 2, 2025
Published: Dec 5, 2025
Copyright
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yadav, V. K., Fang, B., Koomen, J. M., Srivastava, J. and Premi, S. (2025). Quantitative Proteomics of Nitrosylated Proteins in Melanoma Using the Biotin-Switch Technique Combined With Tandem Mass Tag Labeling. Bio-protocol 15(23): e5535. DOI: 10.21769/BioProtoc.5535.
Category
Biochemistry > Protein > Quantification
Systems Biology > Proteomics
Biological Sciences > Biological techniques > Mass spectrometry
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.