- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

A Cytosine Deaminase–Based Genomic Footprinting Assay (cFOOT-seq) for Detecting Transcription Factor Occupancy
(*contributed equally to this work) Published: Vol 16, Iss 6, Mar 20, 2026 DOI: 10.21769/BioProtoc.5637 Views: 23
Reviewed by: Alba BlesaGuohao HanAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2025

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols


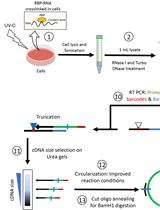
Abstract
Transcription factors (TFs) regulate gene expression by binding to cis-regulatory elements in the genome. Understanding transcriptional regulation requires genome-wide characterization of TF occupancy across different chromatin contexts, yet simultaneous assessment of TF binding for multiple factors remains technically challenging. Here, we describe a detailed and reproducible protocol for cFOOT-seq, a cytosine deaminase–based genomic footprinting assay by sequencing, which enables antibody-independent, base-resolution profiling of chromatin accessibility, nucleosome organization, and TF occupancy. In cFOOT-seq, the double-stranded DNA (dsDNA) cytosine deaminase SsdAtox converts cytosine to uracil in accessible chromatin, whereas TF binding and nucleosome occupancy locally protect DNA from deamination. Using the FootTrack analysis framework, deamination patterns generated by cFOOT-seq are quantitatively analyzed to derive standardized footprint and chromatin organization profiles at base resolution across the genome. Because cFOOT-seq preserves genomic DNA integrity during deamination-based footprinting, it is compatible with ATAC-seq-based chromatin enrichment. ATAC-combined implementations reduce sequencing depth requirements and improve scalability for footprint-focused analyses, supporting applications in low-input and single-cell settings. This protocol provides a practical framework for genome-wide TF footprint profiling and can be readily applied to dissect gene regulatory mechanisms in development, immunity, and disease, including cancer.
Key features
• cFOOT-seq provides antibody-independent, base-resolution, genome-wide profiling of TF occupancy together with chromatin accessibility and nucleosome organization in a single assay.
• Using the FootTrack analysis framework, cFOOT-seq enables quantitative and comparative analysis of TF footprint patterns and occupancy across the genome.
• cFOOT-seq preserves genomic DNA integrity during deamination-based footprinting and is compatible with ATAC-seq-based chromatin enrichment.
• Integrated with ATAC, cFOOT-seq enables sensitive and in-depth TF footprint analyses at low sequencing cost, supporting single-cell applications.
Keywords: Gene transcriptionGraphical overview
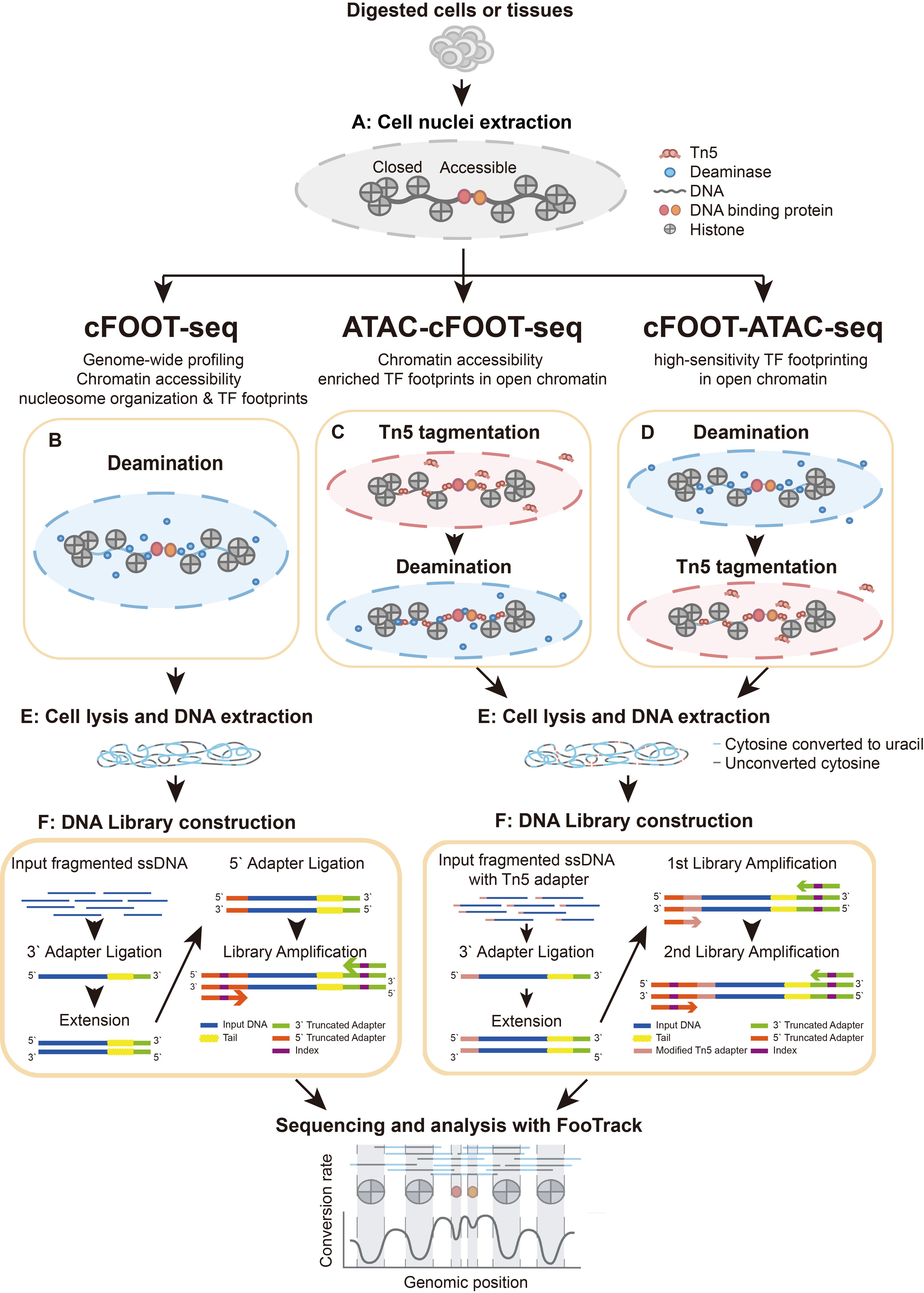
Overview of cFOOT-seq, ATAC-cFOOT-seq, and cFOOT-ATAC-seq procedure
Background
Gene regulation is often mediated by the coordinated activity of multiple transcription factors (TFs) acting at cis-regulatory elements across the genome [1,2]. At cis-regulatory elements, TF occupancy provides a direct readout of where regulatory interactions occur on chromatin. Understanding transcriptional regulation therefore requires approaches that can capture genome-wide TF occupancy information for many TFs in a comparable and integrated manner [3]. However, experimental strategies that enable such large-scale TF occupancy profiling remain limited.
Antibody-based methods for profiling TF binding, including ChIP-seq [4], CUT&RUN [5], CUT&Tag [6–9], DiMeLo-seq [10], and nanoHiMe-seq [11], directly measure protein–DNA interactions in chromatin but depend on antibody availability and specificity and typically profile one TF at a time. This limits scalability and makes it difficult to compare occupancy patterns across multiple TFs within the same sample. TF occupancy can also be inferred computationally from chromatin accessibility data generated by DNase-seq [12,13] or ATAC-seq [14,15]. Although these footprinting approaches are antibody-independent, their sensitivity is influenced by enzyme sequence bias, sequencing depth, and local cutting frequency, and they rely on population-averaged signals rather than directly encoding TF occupancy at the level of individual DNA molecules [16–18].
Single-molecule enzymatic footprinting strategies overcome these limitations by recording protein–DNA interactions as protection patterns against enzymatic modification of DNA. These include methyltransferase-based methods (e.g., NOMe-seq [19], SMF [20], SMAC-seq [21], and Fiber-seq [22]), as well as a growing number of recently reported approaches based on double-stranded DNA (dsDNA) cytosine deaminases, including FOODIE [23], cFOOT-seq [24], TDAC-seq [25], DAF-seq [26], and ACCESS-ATAC [27]. In deaminase-based footprinting, cytosine bases in accessible DNA are converted to uracil, whereas DNA regions occupied by TFs or nucleosomes are protected from deamination. After PCR amplification and sequencing, these protection patterns can be decoded as base-resolution footprints.
cFOOT-seq (a cytosine deaminase-based genomic footprinting assay by sequencing) implements this strategy using SsdAtox [28], a dsDNA cytosine deaminase with robust activity, low sequence bias, and minimal sensitivity to cytosine methylation [24]. This enables antibody-independent, base-resolution profiling of chromatin accessibility, nucleosome organization, and TF occupancy across the genome. Because cFOOT-seq preserves genomic DNA integrity during deamination-based footprinting, it can be combined with ATAC-seq. ATAC-integrated cFOOT-seq workflows enable footprint-focused analyses with reduced sequencing depth and support applications in low-input and single-cell settings. This protocol describes the experimental procedures and the FootTrack-based data analysis framework for cFOOT-seq and its ATAC-integrated workflows.
Materials and reagents
Biological materials
1. K562 (Procell, catalog number: CL-0130)
2. HepG2 (Procell, catalog number: CL0103)
Reagents
1. 1 M Tris-HCl, pH 7.4 (Bioworld, catalog number: 21420063-1)
2. 1 M Tris-HCl, pH 7.5 (Thermo Scientific, catalog number: 15567027)
3. 5 M sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S5150)
4. 0.5 M ethylene diamine tetraacetic acid (EDTA) (Thermo Scientific, catalog number: 15575020)
5. Bovine serum albumin (BSA) (Sigma-Aldrich, catalog number: B2064)
6. 10% Tween-20 (Sigma-Aldrich, catalog number: 655206-50ML)
7. 10% NP-40 (Thermo Scientific, catalog number: 85124)
8. 5% digitonin (Thermo Scientific, catalog number: BN2006)
9. 1 M magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: 63069)
10. COmpleteTM EDTA-free protease inhibitor cocktail (PIC) (Sigma-Aldrich, catalog number: 04693132001)
11. Dimethylformamide (DMF) (Sigma-Aldrich, catalog number: 227056)
12. Ultrapure DNase/RNase-free distilled water (Thermo Scientific, catalog number: 10977023)
13. Dulbecco's phosphate-buffered saline (DPBS) (Gibco, catalog number: C14190500CP)
14. SsdAtox (DNA cytosine deaminase) (in-house purified; aliquot and store at -80 °C)
15. Uracil glycosylase inhibitor (UGI) (New England Biolabs, catalog number: M0281L)
16. Tn5 transposase (tagmentation enzyme; example used: TruePrep® Tagment Enzyme kit, Vazyme, catalog number: S111)
Note: Equivalent Tn5 enzymes from other suppliers can be used. Follow the manufacturer’s instructions for adapter assembly, and a small-scale test is recommended when switching reagents.
17. Lambda DNA (Thermo Scientific, catalog number: SD0021)
18. Glycogen (Sigma-Aldrich, catalog number: 10901393001)
19. Phenol:chloroform:isoamyl alcohol 25:24:1, pH ≥ 7.8 (PCI) (JSENB, catalog number: JS0700-100ML)
20. 100% ethanol (Sinopharm Chemical Reagent, catalog number: 10009218)
21. Isopropanol (Sinopharm Chemical Reagent, catalog number: 40064360)
22. Sodium bicarbonate (NaHCO3) (Sigma-Aldrich, catalog number: S5761-500G)
23. 3 M Sodium acetate buffer solution (NaAc) (Sigma-Aldrich, catalog number: S7899)
24. QubitTM ssDNA Assay kit (Thermo Scientific, catalog number: Q10212)
25. QubitTM dsDNA HS Assay kit (Thermo Scientific, catalog number: Q32854)
26. ssDNA Methylation-Style Library Preparation kit (example used: EpiArt DNA Methylation Library kit for Illumina V3, Vazyme, catalog number: NE103)
Note: Other kits with the same functionality (ssDNA library construction and compatibility with deaminated DNA templates) can also be used. A small-scale test is recommended to confirm library yield and fragment-size distribution.
27. DNA clean-up magnetic beads (SPRI-based) (example used: VAHTS DNA Clean Beads, Vazyme, catalog number: N411)
Note: Equivalent SPRI beads can be substituted. A small-scale test is recommended to confirm recovery and size selection performance.
28. Oligonucleotides for Tn5 transposome generation and library preparation (listed in Table 1)
Table 1. Sequences of oligonucleotides used in this protocol
| Name | Sequence (5′→3′) | Purpose | Notes/source |
|---|---|---|---|
| Tn5 primer D | TGGTAGAGAGGGTGAGATGTGTATAAGAGACAG | Cytosine-free Tn5 adapter (top strand) | Custom oligo; lacks cytosines |
| Tn5 ME oligo | phos-CTGTCTCTTATACACATCT-NH2 | Universal Tn5 mosaic end | Modified 5′ phosphate, 3′ NH2 |
| i5_bridge primer | ACACTCTTTCCCTACACGACGCTCTTCCGATCTTGGTAGAGAGGGTGAGATGTGTATAAGAGACAG | Add 5′ sequencing handle during pre-amplification | Custom oligo |
| Universal i7 primer | Provided in index primer kit | PCR amplification and indexing | Illumina-compatible i7 index primer |
| Universal i5 primer | Provided in index primer kit | PCR amplification and indexing | Illumina-compatible i5 index primer |
Solutions
1. 10% BSA in H2O (see Recipes)
2. 10% BSA in DPBS (see Recipes)
3. Transposome generation (see Recipes)
4. Nuclei extraction buffer (see Recipes)
5. Stop buffer (see Recipes)
6. 2× SsdAtox reaction buffer (2× SRB) (see Recipes)
7. SsdAtox-UGI mix (see Recipes)
8. Cell suspension buffer (see Recipes)
9. Hot lysis buffer (see Recipes)
10. 2× TD buffer (see Recipes)
11. Transposition reaction mix (see Recipes)
12. Tn5 washing buffer (see Recipes)
13. ATAC-cFOOT-cell suspension buffer (see Recipes)
14. 2× SsdAtox reaction buffer plus DPBS and BSA (2× SRB+DB) (see Recipes)
15. cFOOT-ATAC-SsdAtox-UGI mix (see Recipes)
16. cFOOT-ATAC-cell suspension buffer (see Recipes)
17. Deaminase washing buffer (see Recipes)
Recipes
1. 10%BSA in H2O
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| Ultrapure DNase/RNase-free distilled water | N/A | Up to 10 mL |
| BSA powder | 10% (w/v) | 1 g |
| Total | N/A | 10 mL |
Before use, filter the solution through the Millex®-HV filter unit. This buffer should be stored at -20 °C.
2. 10% BSA in DPBS
| Reagent | Final concentration | Quantity or volume |
|---|---|---|
| DPBS | N/A | Up to 10 mL |
| BSA powder | 10% (w/v) | 1 g |
| Total | N/A | 10 mL |
Before use, filter the solution through the Millex®-HV filter unit. This buffer should be stored at -20 °C.
3. Transposome generation
Transposome assembly was performed using a Tn5-based workflow adapted from standard tagmentation procedures (example used: TruePrep® Tagment Enzyme kit, Vazyme). Tn5 transposase from other suppliers can also be used. However, the cytosine-free adapters described below should be used to avoid cytosine deamination by SsdAtox.
The adapter was generated by annealing Tn5 Primer D (lacking cytosines in its 5′ overhang) with the universal Tn5 ME oligo. The annealed adapter was then loaded onto Tn5 transposase to form the active transposome, following the manufacturer’s recommended annealing and loading instructions for the chosen Tn5 system. The assembled transposome can be aliquoted and stored at -20 °C (avoid repeated freeze–thaw cycles).
4. Nuclei extraction buffer
| Reagent | Final concentration | Volume for 10 reactions |
|---|---|---|
| 1 M Tris-HCl, pH 7.4 | 10 mM | 5 μL |
| 5 M NaCl | 10 mM | 1 μL |
| 0.1 M EDTA | 0.1 mM | 0.5 μL |
| 10% BSA in H2O | 1% | 50 μL |
| 10% Tween-20 | 0.1% | 5 μL |
| 10% NP-40 | 0.1% | 5 μL |
| 5% Digitonin | 0.01% | 1 μL |
| 1 M MgCl2 | 3 mM | 1.5 μL |
| 50× PIC | 1× | 10 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 421 μL |
| Total | N/A | 500 μL |
Prepare immediately before use and keep at 0–4 °C until use. The commercially available EDTA solution is 0.5 M and should be diluted to 0.1 M for use as a stock solution when preparing the nuclei extraction buffer. Optional: The nuclei extraction buffer without PIC and any detergents can be prepared ahead of time and stored at -20 °C for up to 1 month. Add the detergents and PIC immediately before use.
5. Stop buffer
| Reagent | Final concentration | Volume for 10 reactions |
|---|---|---|
| 1 M Tris-HCl, pH 7.4 | 10 mM | 100 μL |
| 5 M NaCl | 10 mM | 20 μL |
| 0.1 M EDTA | 0.1 mM | 10 μL |
| 10% BSA in H2O | 1% | 1 mL |
| Ultrapure DNase/RNase-free distilled water | N/A | 8870 μL |
| Total | N/A | 10 mL |
Prepare immediately before use and keep at 0–4 °C until use.
6. 2× SsdAtox reaction buffer (2× SRB)
| Reagent | Final concentration | Volume for 10 reactions |
|---|---|---|
| 1 M Tris-HCl, pH 7.4 | 20 mM | 5 μL |
| 0.1 M DTT | 2 mM | 5 μL |
| 50× PIC | 2× | 10 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 230 μL |
| Total | N/A | 250 μL |
Prepare immediately before use and keep at 0–4 °C until use.
7. SsdAtox-UGI mix
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× SRB | 1× | 10 μL |
| 2 U/μL UGI | 0.1 U/μL | 2.5 μL |
| SsdAtox | 3–20 U/μL | × μL |
| Ultrapure DNase/RNase-free distilled water | N/A | Up to 20 μL |
Prepare immediately before use and keep at 0–4 °C until use.
8. Cell suspension buffer
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× SRB | 1× | 15 μL |
| 0.4 ng/μL Lambda DNA | 0.04 ng/μL | 5 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | Up to 30 μL |
Prepare immediately before use and keep at 0–4 °C until use.
9. Hot lysis buffer
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 1 M NaHCO3 | 100 mM | 25 μL |
| 10% SDS | 1% | 5 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 150 μL |
| Total | N/A | 200 μL |
Prepare immediately before use and keep at room temperature.
10. 2× TD buffer
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 1 M Tris-HCl, pH 7.5 | 20 mM | 0.5 μL |
| 1 M MgCl2 | 10 mM | 0.25 μL |
| 100% DMF | 20% | 5 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 19.25 μL |
| Total | N/A | 25 μL |
Prepare immediately before use and keep at 0–4 °C until use.
11. Transposition reaction mix
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× TD buffer | 1× | 25 μL |
| DPBS | 33% (v/v) | 16.5 μL |
| Transposase | 100 nM | 0.667 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 7.833 μL |
| Total | N/A | 50 μL |
Prepare fresh on ice immediately before use and keep at 0–4 °C until use.
12. Tn5 washing buffer
| Reagent | Final concentration | Volume for 1× reaction |
|---|---|---|
| 2× SRB | 1× | 250 μL |
| 0.1 M EDTA | 454.5 nM | 2.5 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 247.5 μL |
| Total | N/A | 500 μL |
Prepare immediately before use and keep at 0–4 °C until use.
13. ATAC-cFOOT-cell suspension buffer
| Reagent | Final concentration | Volume for 1× reaction |
|---|---|---|
| 2× SRB | 1× | 15 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 15 μL |
| Total | N/A | 30 μL |
Prepare immediately before use and keep at 0–4 °C until use.
14. 2× SsdAtox reaction buffer plus DPBS and BSA (2× SRB+DB)
| Reagent | Final concentration | Volume for 10 reactions |
|---|---|---|
| 1 M Tris-HCl, pH 7.4 | 20 mM | 5 μL |
| 0.1 M DTT | 2 mM | 5 μL |
| DPBS | 66% (v/v) | 165 μL |
| 10% BSA in H2O | 0.04% | 1 μL |
| 50× PIC | 2× | 10 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 64 μL |
| Total | N/A | 250 μL |
Note: We typically prepare 2× SRB+DB fresh (with DTT and PIC) and keep it at 0–4 °C until use. Optional: A 2× SRB+DB base without DTT and PIC can be prepared in advance, aliquoted, and stored at -20 °C for up to 1 month. Add DTT and PIC immediately before use.
15. cFOOT-ATAC-SsdAtox -UGI mix
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× SRB+DB | 1× | 10 μL |
| 2 U/μL UGI | 0.1 U/μL | 2.5 μL |
| SsdAtox | N/A | × μL |
| Ultrapure DNase/RNase-free distilled water | N/A | Up to 20 μL |
Prepare immediately before use and keep at 0–4 °C until use.
16. cFOOT-ATAC-cell suspension buffer
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× SRB+DB | 1× | 15 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 15 μL |
| Total | N/A | 30 μL |
Prepare immediately before use and keep at 0–4 °C until use.
17. Deaminase washing buffer
| Reagent | Final concentration | Volume per reaction (1×) |
|---|---|---|
| 2× SRB+DB | 1× | 250 μL |
| Ultrapure DNase/RNase-free distilled water | N/A | 250 μL |
| Total | N/A | 500 μL |
Prepare fresh immediately before use and keep at 0–4 °C until use.
Note: Before the experiment, pre-mix well every buffer above.
Laboratory supplies
1. 1.5 mL DNase/RNase-free tubes (Axygen, catalog number: MCT-150-C)
2. 0.2 mL DNase/RNase-free PCR tubes (Axygen, catalog number: PCR-02-C)
3. 50 mL conical tubes (Biobest, catalog number: BSD-HCT-50A)
4. 15 mL conical tubes (Biobest, catalog number: BSD-HCT-15A)
5. 1.5 mL DNA LoBind® tube (Eppendorf, catalog number: 0030108051)
6. 10 μL filter pipette tips (Biobest, catalog number: BSD-HTF-CTL-10)
7. 20 μL filter pipette tips (Biofil, catalog number: PMT252520)
8. 100 μL filter pipette tips (Biofil, catalog number: PMT252100)
9. 200 μL filter pipette tips (Biofil, catalog number: PMT231200)
10. 1 mL filter pipette tips (Biobest, catalog number: BSD-HTF-1250-AR)
11. Millex®-HV filter unit (Millipore, catalog number: SLHV033RB)
12. microTUBE-50 AFA fiber screw-cap case (Covaris, catalog number: 520166)
Equipment
1. FinnpipetteTM F1 variable volume single-channel pipettes, 100–1,000 μL (Thermo Scientific, catalog number: 4641100N)
2. FinnpipetteTM F1 variable volume pipettes, 20–200 μL (Thermo Scientific, catalog number: 4641080N)
3. FinnpipetteTM F1 variable volume pipettes, 10–100 μL (Thermo Scientific, catalog number: 4641070N)
4. FinnpipetteTM F1 variable volume pipettes, 2–20 μL (Thermo Scientific, catalog number: 4641060N)
5. FinnpipetteTM F1 variable volume pipettes, 1–10 μL (Thermo Scientific, catalog number: 4641030N)
6. FinnpipetteTM F1 variable volume pipettes, 0.2–2 μL (Thermo Scientific, catalog number: 4641010N)
7. Thermomixer (Yeasen, catalog number: 80440ES03)
8. QubitTM 4 NGS Starter Kit, with WiFi (Thermo Scientific, catalog number: Q33240)
9. Refrigerated centrifuge (Eppendorf, model: 5425 R)
10. M220 Focused ultrasonicator (Covaris, catalog number: 500295)
11. T100 thermal cycler (Bio-Rad, catalog number: 1861096)
Software and datasets
1. FastQC (v0.12.1): Quality control of raw sequencing reads, including assessment of base quality scores, GC content, sequence duplication levels, and overrepresented sequences. Source: https://www.bioinformatics.babraham.ac.uk/projects/fastqc
2. Trim Galore (v0.6.10): Adapter trimming and removal of low-quality bases from sequencing reads. Source: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore
3. BASAL (v1.8): Conversion-aware mapping of nucleotide base-conversion sequencing reads. Source: https://github.com/JiejunShi/BASAL
4. BasalKit (v1.8): Extraction of per-base sequencing coverage and C-to-T conversion rates from BASAL-aligned BAM files, followed by generation of base-conversion tables for downstream analysis. BasalKit is an integrated utility within the BASAL framework specifically designed for base-conversion sequencing data. Source: https://github.com/JiejunShi/BASAL (distributed as part of the BASAL package)
5. bedtools (v2.31.1): Removal of genomic positions overlapping known single-nucleotide polymorphisms (SNPs) to prevent interference with the accurate estimation of base-conversion rates. Source: https://bedtools.readthedocs.io/en/latest
6. FootTrack (v1.1.0): Comprehensive analysis of transcription factor occupancy from conversion-based sequencing data, including bias correction, footprint scoring, differential TF binding analysis, and de novo TF binding site prediction. Source: https://github.com/ZhangLab-TJ/FootTrack
7. ATAC-seq and transcription factor ChIP-seq datasets used in this protocol were primarily obtained from the ENCODE Project and the Cistrome Project (https://www.encodeproject.org; http://cistrome.org). Detailed descriptions of the datasets, including experiment identifiers and accession numbers, can be found in the original article [24]. Transcription factor motif files were downloaded from the JASPAR database (https://jaspar.genereg.net) and used for genome-wide motif scanning and footprint-based TF binding analysis.
Single-nucleotide polymorphism data for mouse genomes were obtained from the Mouse Genomes Project (https://www.mousegenomes.org/snps-indels). SNP information for human cell lines was retrieved from the ENCODE Project (https://www.encodeproject.or/) and used to mask polymorphic positions during base-conversion rate calculations.
Procedure
A. Cell nuclei extraction
1. Digest the tissue or cell line of interest and prepare a single-cell suspension.
Note: If you need to use tissue cells, it is recommended that users first try to detect the total number of cells after the tissue to be tested is digested into a single cell. According to experimental needs, we can get enough cells according to the proportion of 50,000 cells for each experimental group.
2. Prewash 1.5 mL of DNase/RNase-free tubes with 1 mL of DPBS containing 5% BSA (prepared by diluting 10% BSA in DPBS). Then, remove all DPBS containing 5% BSA. Transfer 50,000 cells into each tube and centrifuge at 300× g for 5 min at 4 °C.
3. Aspirate all supernatant and wash cells once with 100 μL of DPBS containing 0.04% BSA (prepared by diluting 10% BSA in DPBS). Centrifuge at 300× g for 5 min at 4 °C.
4. Carefully aspirate all supernatant, avoiding disturbing the cell pellet. Gently flick the bottom of the tube with your finger to loosen and disperse the pellet.
5. Add ice-cold nuclei extraction buffer at a ratio of 50 μL of buffer per 50,000 cells. Mix thoroughly by pipetting up and down 3 times.
6. Incubate on ice for 3 min, then immediately add 1 mL of stop buffer to each tube, close the caps, and gently invert the tubes up and down 5 times to stop nuclei extraction.
7. Centrifuge at 700× g for 5 min at 4 °C.
8. Carefully aspirate all supernatant, avoiding disturbing the cell pellet.
CRITICAL: To minimize residual liquid, aspirate most of the supernatant with a 1 mL pipette, leaving approximately 40–60 μL above the pellet. Centrifuge again at 700× g for 2 min at 4 °C, then remove the remaining supernatant with a 100 μL pipette.
CAUTION: When starting from ~50,000 cells, the nuclei pellet is usually visible as a small white pellet before permeabilization. After permeabilization, the pellet may become transparent. When aspirating, avoid touching the tube bottom and leave a small residual volume if needed to prevent pellet loss.
9. Gently flick the bottom of the tube with your finger to loosen the nuclei pellet.
10. Proceed with one of the following workflows:
a. If proceeding to cFOOT-seq, continue with section B.
b. If proceeding to ATAC–cFOOT-seq, continue with section C.
c. If proceeding to cFOOT–ATAC-seq, continue with section D.
Note: After completing the selected workflow (section B, C, or D), proceed to section E.
B. Enzyme reaction for cFOOT-seq
1. Add 30 μL of ice-cold cell suspension buffer to each tube and resuspend the nuclei thoroughly by pipetting up and down 5 times.
2. Add 20 μL of ice-cold SsdAtox-UGI mix and mix well by pipetting up and down 5 times.
3. Place the tubes in a thermomixer and incubate at 37 °C with shaking at 1,000 rpm for 10 min.
CRITICAL: This step is highly sensitive to reaction temperature and incubation time. It is essential to ensure that the reaction time is strictly consistent across all samples. If multiple experimental groups are processed in parallel, tubes can be kept on ice after adding the SsdAtox–UGI mix and then transferred simultaneously to a thermomixer preheated to 37 °C to initiate all reactions at the same time.
4. Immediately place the tubes on ice to stop the reaction.
C. Enzyme reaction for ATAC-cFOOT-seq
1. Add 50 μL of ice-cold transposition reaction mix to each tube and resuspend the nuclei by pipetting up and down 5 times.
2. Place the tubes in a thermomixer and incubate at 37 °C with shaking at 1,000 rpm for 15 min.
3. Add 500 μL of ice-cold Tn5 washing buffer to each tube and mix by incubating at 37 °C with shaking at 1,000 rpm for 2 min.
4. Centrifuge at 700× g for 5 min at 4 °C.
5. Carefully remove the supernatant without disturbing the nuclei pellet and gently flick the bottom of the tube to loosen the pellet.
CRITICAL: To minimize residual liquid, aspirate most of the supernatant with a 1 mL pipette, leaving approximately 40–60 μL above the pellet. Centrifuge again at 700× g for 2 min at 4 °C, then remove the remaining supernatant with a 100 μL pipette.
6. Add 30 μL of ice-cold ATAC-cFOOT-cell suspension buffer to each tube and resuspend the nuclei thoroughly by pipetting up and down 5 times.
7. Add 20 μL of ice-cold SsdAtox-UGI mix and mix well by pipetting up and down 5 times.
CRITICAL: This step is highly sensitive to reaction temperature and incubation time. It is essential to ensure that the reaction time is strictly consistent across all samples. If multiple experimental groups are processed in parallel, tubes can be kept on ice after adding the SsdAtox–UGI mix and then transferred simultaneously to a thermomixer preheated to 37 °C to initiate all reactions at the same time.
8. Place the tubes in a thermomixer and incubate at 37 °C with shaking at 1,000 rpm for 10 min.
9. Immediately place the tubes on ice to stop the reaction.
D. Enzyme reaction for cFOOT-ATAC-seq
1. Add 30 μL of ice-cold cFOOT-ATAC-cell suspension buffer to each tube and resuspend the nuclei thoroughly by pipetting up and down 5 times.
2. Add 20 μL of ice-cold cFOOT-ATAC-SsdAtox-UGI mix and mix well by pipetting up and down 5 times.
3. Place the tubes in a thermomixer and incubate at 37 °C with shaking at 1,000 rpm for 10 min.
4. Immediately place the tubes on ice. Add 500 μL of ice-cold deaminase washing buffer to each tube, return the tubes to the thermomixer, and shake at 1,000 rpm for 2 min at 37 °C to mix the nuclei with the washing buffer.
5. Centrifuge at 700× g for 5 min at 4 °C.
6. Carefully remove the supernatant without disturbing the nuclei pellet and gently flick the bottom of the tube to loosen the pellet.
CRITICAL: To minimize residual liquid, aspirate most of the supernatant with a 1 mL pipette, leaving approximately 40–60 μL above the pellet. Centrifuge again at 700× g for 2 min at 4 °C, then remove the remaining supernatant with a 100 μL pipette.
7. Add 50 μL of ice-cold transposition reaction mix to each tube and resuspend the nuclei by pipetting up and down 5 times.
8. Incubate the tubes in a thermomixer at 37 °C with shaking at 1,000 rpm for 15 min.
9. Immediately place the tubes on ice to stop the reaction.
E. Cell lysis and DNA purification
1. Add 200 μL of hot lysis buffer to each 50 μL reaction and incubate the tubes in a thermomixer at 1,350 rpm for 90 min at 65 °C.
2. Add an equal volume (250 μL) of phenol:chloroform:isoamyl alcohol 25:24:1 (pH ≥ 7.8) to each tube. Vortex vigorously for 15 s to mix the organic and aqueous phases. Centrifuge the tubes in a benchtop high-speed centrifuge at 15,000 rpm for 5 min at room temperature, then carefully transfer the aqueous phase (topper phase) to a new DNA LoBind tube.
3. Add 1/10 volume of 3 M NaAc and 2 μL of glycogen to each tube. Vortex to mix and then add an equal volume of isopropanol and vortex again.
4. Place the samples at -80 °C overnight.
SAFE STOP POINT: At this step, the procedure can be paused for up to 1 month. The samples are stable at -80 °C.
5. Remove the samples from the freezer and thaw completely at room temperature. Centrifuge at 20,000× g for 20 min at 4 °C.
6. Remove the supernatant as in step A4, taking care not to disturb the DNA pellet. Add 800 μL of ice-cold 80% (v/v) ethanol, gently invert the tubes 5 times to wash the pellet, incubate the tubes on ice for 5 min, and then centrifuge at 20,000× g for 5 min at 4 °C.
7. Repeat the DNA ethanol wash described in step E6 once.
8. Remove the supernatant as completely as possible without disturbing the DNA pellet. Open the tube caps and air-dry the pellet until it becomes semi-translucent.
9. Add 20 μL of ultrapure water to dissolve the DNA.
Note: Pipette the solution up and down several times after adding water to facilitate complete dissolution of the DNA pellet. With an input of 50,000 cells, the total amount of DNA obtained is generally between 200 and 300 ng.
F. DNA library construction
DNA library construction differs depending on the selected workflow.
F1. Library construction for cFOOT-seq
1. Dilute 100 ng of genomic DNA with ultrapure DNase/RNase-free distilled water to a final volume of 50 μL.
2. Transfer the diluted DNA to a microTUBE-50 and shear using a Covaris M220 sonicator with the following parameters: peak incident power: 175 W; duty factor: 10%; cycles per burst: 200; treatment time: 40 s; temperature: 7 °C.
3. Use 20 ng of DNA for library preparation. Construct libraries using a commercially available ssDNA methylation-style library preparation kit for Illumina (e.g., EpiArt DNA Methylation Library Kit, Vazyme) according to the manufacturer’s instructions. See General note 4 for quality control of DNA library construction.
Note: Other functionally equivalent ssDNA library preparation kits compatible with uracil-containing (deaminated) DNA may also be used. We recommend a small-scale pilot to confirm library yield and fragment-size distribution.
F2. Library construction for ATAC-cFOOT-seq or cFOOT-ATAC-seq
1. Denature 50 ng of genomic DNA, ligate truncated adapters at the 3′ ends, and perform strand extension to generate double-stranded DNA using an ssDNA methylation-style library preparation kit for Illumina (e.g., EpiArt DNA Methylation Library Kit, Vazyme). Purify the DNA using 1× VAHTS DNA clean beads.
2. Perform four cycles of initial amplification using the universal i7 primer and i5_bridge primer to add the 5′ sequencing handle. Purify the PCR products using 1× VAHTS DNA clean beads.
3. Perform eight additional cycles of amplification using universal i5 and i7 primers from a standard Illumina-compatible multiplex oligo set to generate sequencing-ready libraries.
4. Size-select 300–500 bp fragments by agarose gel electrophoresis to obtain the final sequencing library. See General note 4 about quality control for DNA library construction.
Note: Reagents listed above are examples used. Other functionally equivalent ssDNA library preparation kits compatible with uracil-containing (deaminated) DNA, as well as SPRI-based DNA clean beads, can also be used. We recommend a small-scale pilot study to confirm library yield and fragment-size distribution.
H. Sequencing
1. Sequence libraries using paired-end 150 bp reads on an Illumina NovaSeq 6000 system. Select the corresponding sequencing depth for the appropriate workflow according to Table 2.
Table 2. Recommended sequencing depth and effective coverage for different cFOOT-based workflows.
| Workflow | Analysis objective | Read pairs per sample | Effective coverage |
| cFOOT-seq | Condition testing | ~7 million | ~1.5× genome-wide |
| De novo footprint analysis | ~100 million | ~6.5× genome-wide | |
| ATAC-cFOOT-seq | Condition testing | ~3 million | ~6× in accessible chromatin |
| De novo footprint analysis | ~60 million | ~42× in accessible chromatin | |
| cFOOT-ATAC-seq | Condition testing | ~3 million | ~2× in accessible chromatin |
| De novo footprint analysis | ~60 million | ~13× in accessible chromatin |
Data analysis
Data analysis of cFOOT-seq data is centered on the FootTrack framework, which provides an integrated approach for decoding TF footprints, chromatin accessibility, and nucleosome organization from cytosine deamination patterns at base resolution. The data analysis workflow is shown in Figure 1.
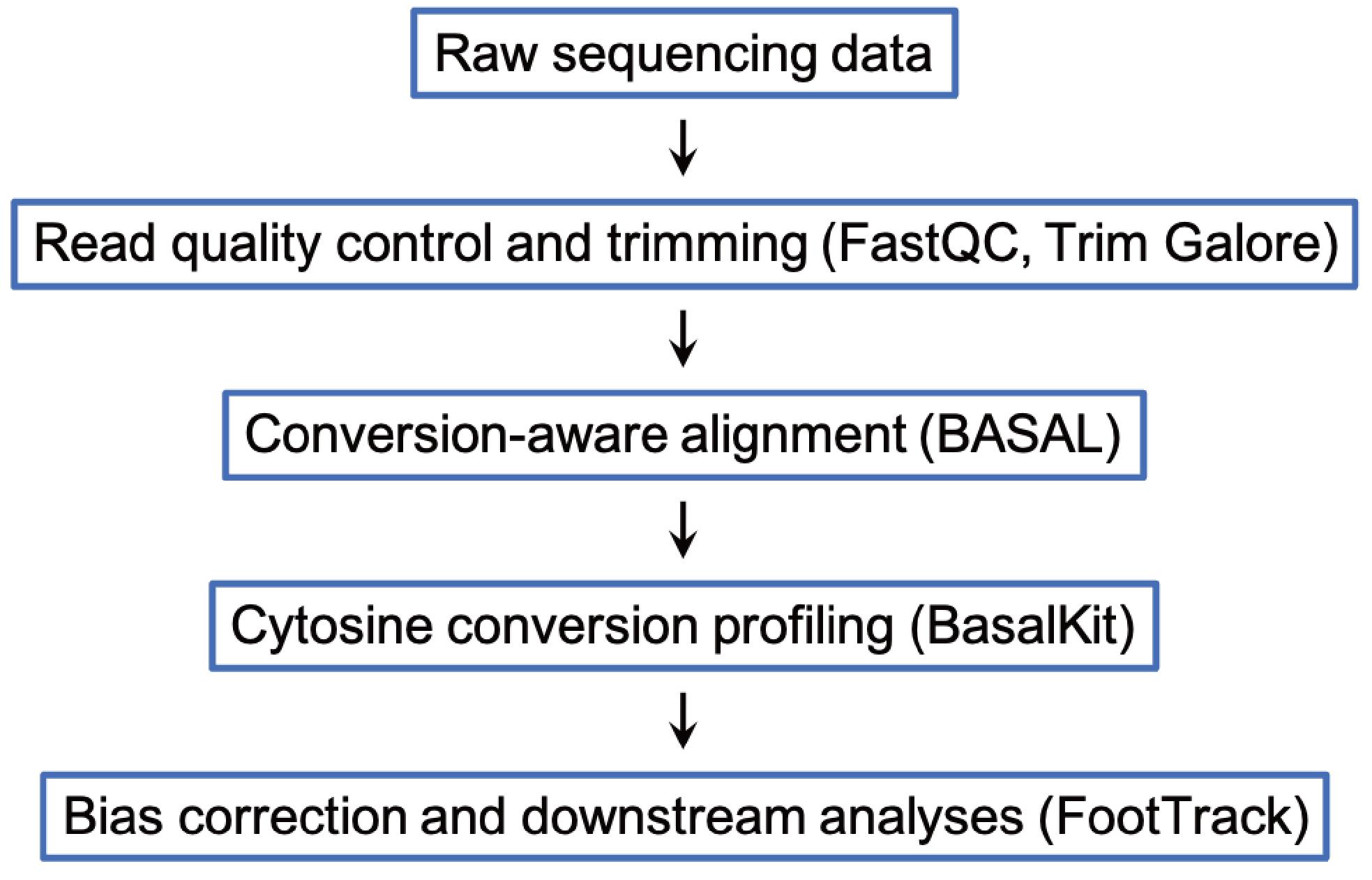
Figure 1. Workflow of data analysis of cFOOT-seq
Raw paired-end sequencing reads were first subjected to quality assessment using FastQC (v0.12.1) and subsequently processed with Trim Galore (v0.6.10) to remove adapter contamination and low-quality bases. For bulk cFOOT-seq libraries, the following command was used to ensure read quality suitable for footprint analysis:
trim_galore --trim-n --clip_R1 3 --clip_R2 10 --three_prime_clip_R1 3 --three_prime_clip_R2 3 --length 35 -q 20 --fastqc --paired $dir/00.rename/${i}.R1.fq.gz $dir/00.rename/${i}.R2.fq.gz -o $dir/01.filterBecause cFOOT-seq libraries are generated using a single-stranded DNA-based protocol, asymmetric trimming parameters were applied (i.e., --clip_R1 3 --clip_R2 10 --three_prime_clip_R1 3 --three_prime_clip_R2 3) to account for strand-specific library structure. These parameters may be adjusted accordingly when alternative library preparation strategies are used.
Trimmed reads were aligned to the appropriate reference genome using BASAL (v1.8), a mapping tool optimized for nucleotide base-conversion sequencing. Alignments were performed in C-to-T conversion mode using the following command:
basal -a $dir/01.filter/${i}.R1_val_1.fq.gz -b $dir/01.filter/${i}.R2_val_2.fq.gz -d ${genome} -m 1 -x 1000 -p ${core} -v 0.06 -n 0 -g 1 -M C:T -o ${i}.bamBecause the library was prepared using a single-stranded protocol, the parameter -n 0 was specified to enforce strand-specific alignment. This setting can be modified for datasets generated using different library construction methods.
Per-cytosine read depth and C-to-T conversion rates were calculated using the BasalKit avgmod module (-r -m 1 -i correct) to generate genome-wide cytosine conversion tables. The following command was used:
python ${basalkit} avgmod $dir/02.align/${i}.sort.bam ${genome} -r -m 1 -o ${i}_basal_methratio -i correctKnown single-nucleotide polymorphisms, as described in the Software and datasets section, were excluded to avoid confounding true TF footprints with polymorphism-driven base changes.
The resulting conversion profiles were analyzed using FootTrack for bias correction, footprint detection, quantitative TF occupancy analysis, and comparative analysis across conditions. Detailed scripts and example pipelines are available at https://github.com/ZhangLab-TJ/FootTrack.
Representative analysis outputs are shown in Figure 2 and serve as reference patterns for data quality assessment. Aggregated conversion profiles around transcription start sites or ATAC-seq-defined open chromatin regions reflect expected accessibility patterns (Figure 2A, B), whereas localized depletion of cytosine conversion at known TF (CTCF) binding sites reveals TF footprints accompanied by phased nucleosome organization (Figure 2C, D).
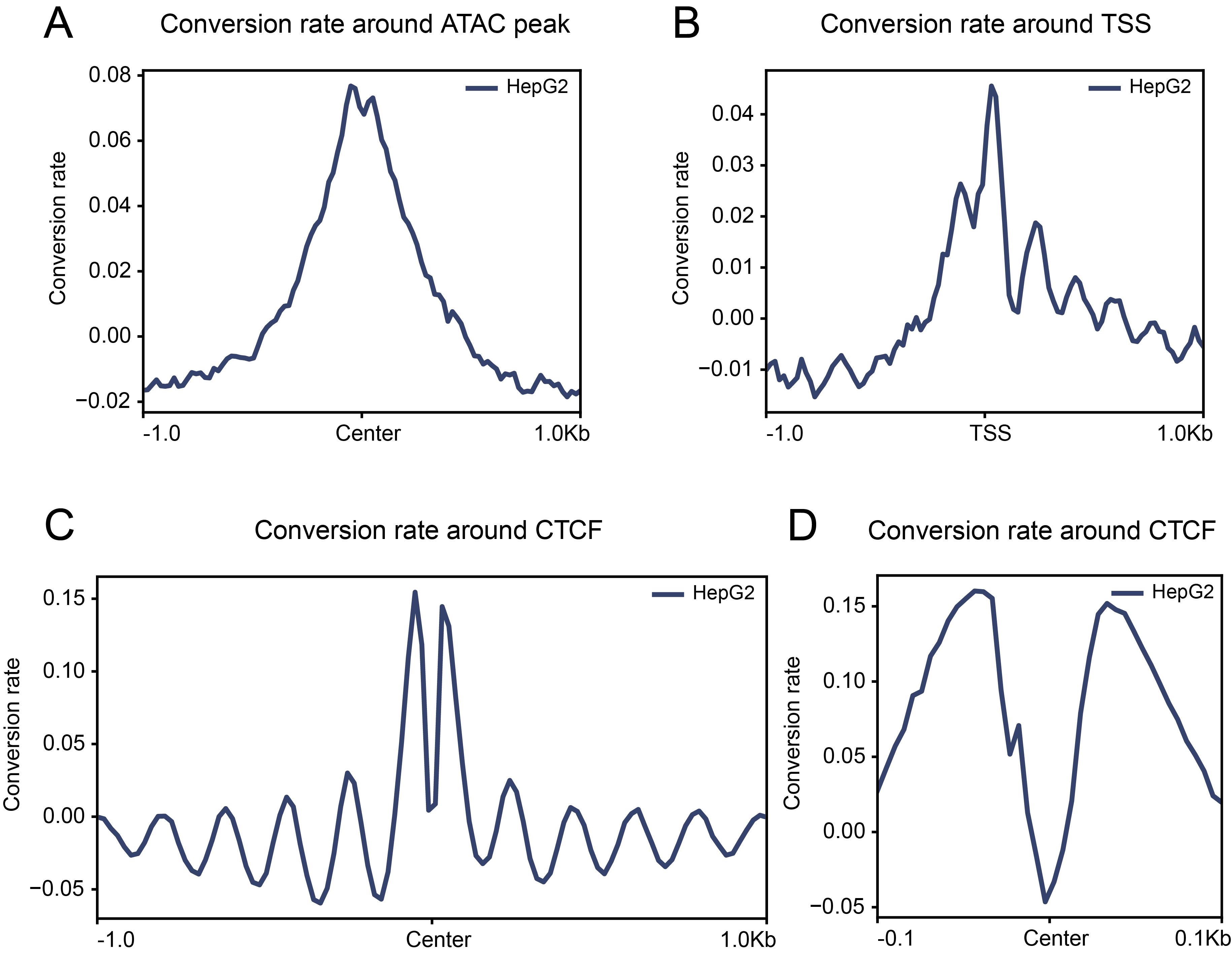
Figure 2. Schematic overview of cFOOT-seq data analysis results. (A) Average normalized DNA conversion rate profiling around the center regions of open chromatin regions (ORCs) defined by ATAC-seq data in HepG2 cells. (B) Average normalized DNA conversion rate profiling around transcription start sites (TSS) in HepG2 cells. (C, D) Average normalized DNA conversion rate profiling around CTCF binding sites defined by ChIP-seq in HepG2 cells, illustrating the position information of nucleosomes around CTCF (C) and footprint of CTCF (D).
Validation of protocol
This protocol (or parts of it) has been used and validated in the following research article:
• Wang et al. [24]. Genome-wide investigation of transcription factor footprints and dynamics using cFOOT-seq. Protein Cell 16(11): 932–952. DOI: 10.1093/procel/pwaf071.
General notes and troubleshooting
General notes
1. Workflow selection: Three workflows are provided in this protocol: standard cFOOT-seq, ATAC-cFOOT-seq, and cFOOT-ATAC-seq. Standard cFOOT-seq provides the most comprehensive information by simultaneously capturing TF occupancy, chromatin accessibility, and nucleosome organization, and is recommended when full chromatin structural information is required. ATAC-integrated workflows (ATAC-cFOOT-seq and cFOOT-ATAC-seq) enrich accessible chromatin regions prior to or following deamination-based footprinting, thereby reducing the sequencing depth required for footprint analysis and lowering overall sequencing cost. Among these, ATAC-cFOOT-seq is recommended for general use, particularly for experiments requiring robust chromatin accessibility measurements, reduced sequencing cost, or compatibility with single-cell applications. The cFOOT-ATAC-seq workflow provides higher sensitivity for TF footprint detection but compromises ATAC enrichment efficiency. This workflow is therefore best suited for specialized, footprint-focused analyses where maximal footprint sensitivity is prioritized. If chromatin accessibility information is required, a separate ATAC-seq experiment is recommended.
2. Cell handling: Although this protocol is demonstrated using the immortal cell lines K562 and HepG2, the cFOOT-seq workflow is not restricted to cell lines and can also be applied to primary cells. For each new cell type, optimization of nuclei permeabilization and SsdAtox input is recommended. cFOOT-seq is sensitive to chromatin state and is performed under native, non-fixed conditions. Chemical fixation is not recommended, as crosslinking reduces deaminase accessibility and compromises footprint detection. Cell handling and lysis conditions should be kept consistent across samples intended for comparison.
3. Enzyme optimization: Because the activity of SsdAtox obtained through protein purification exhibits batch-to-batch variability, we recommend defining enzyme input based on activity units prior to performing cellular titration experiments. Detailed methods for enzyme activity determination are described in [24]. We define one unit of enzyme activity as the minimum amount of enzyme required to completely deaminate a total of 10 pmol of DNA substrate within 30 min in an in vitro reaction under enzymatic conditions at 37 °C. For each new cell type or tissue, a deaminase titration experiment should be performed by treating equal numbers of cells with a gradient of SsdAtox concentrations. The recommended enzyme concentration range is 3–20 U/μL, within which multiple concentrations should be evaluated. After sequencing and data analysis, the enzyme amount yielding a genome-wide cytosine conversion rate of 25%–35% (for cFOOT-seq and cFOOT-ATAC-seq) or 35%–45% (for ATAC-cFOOT-seq) should be selected and used for subsequent experiments.
4. Control of conversion rate variability for comparative analyses: For comparative analyses across different experimental groups (e.g., differential analysis or correlation analysis), the difference in genome-wide conversion rate between samples should not exceed 5%. Larger differences in conversion rate may introduce systematic bias and compromise quantitative comparison of TF footprints across samples. We therefore recommend performing comparisons only among samples in the same batch that pass this criterion and reporting the genome-wide conversion rate for each sample as a QC metric. If the difference exceeds 5%, we suggest re-matching samples (or repeating the experiment) rather than applying batch-effect correction within the current analysis workflow.
5. In-process QC checkpoints during cFOOT-seq: (Before permeabilization) After centrifugation, a visible white pellet should be present at the tube bottom when starting from ~50,000 cells/nuclei. (After permeabilization) The pellet often becomes transparent and difficult to see. When removing supernatant, avoid touching the tube bottom (leave a small residual volume if needed). (After gDNA extraction) Measure gDNA concentration (e.g., Qubit) to confirm successful recovery. (After PCR amplification) Measure library yield (e.g., Qubit) to ensure sufficient amplification.
6. Quality control for DNA library construction
a. For cFOOT-seq: DNA library fragment sizes should predominantly fall within 300–500 bp, with a strong enrichment around ~300 bp (Figure 3A). With 20 ng of input genomic DNA, the final library yield after 7 PCR cycles should be no less than 200 ng.
b. For cFOOT-ATAC-seq or ATAC-cFOOT-seq: DNA library fragment size distribution should exhibit the classic ATAC-seq pattern, and DNA fragment size starts at ~300 bp with distinct peaks occurring every ~150 bp (Figure 3B). With 50 ng of input DNA, the final library yield after 12 PCR cycles should be no less than 300 ng.
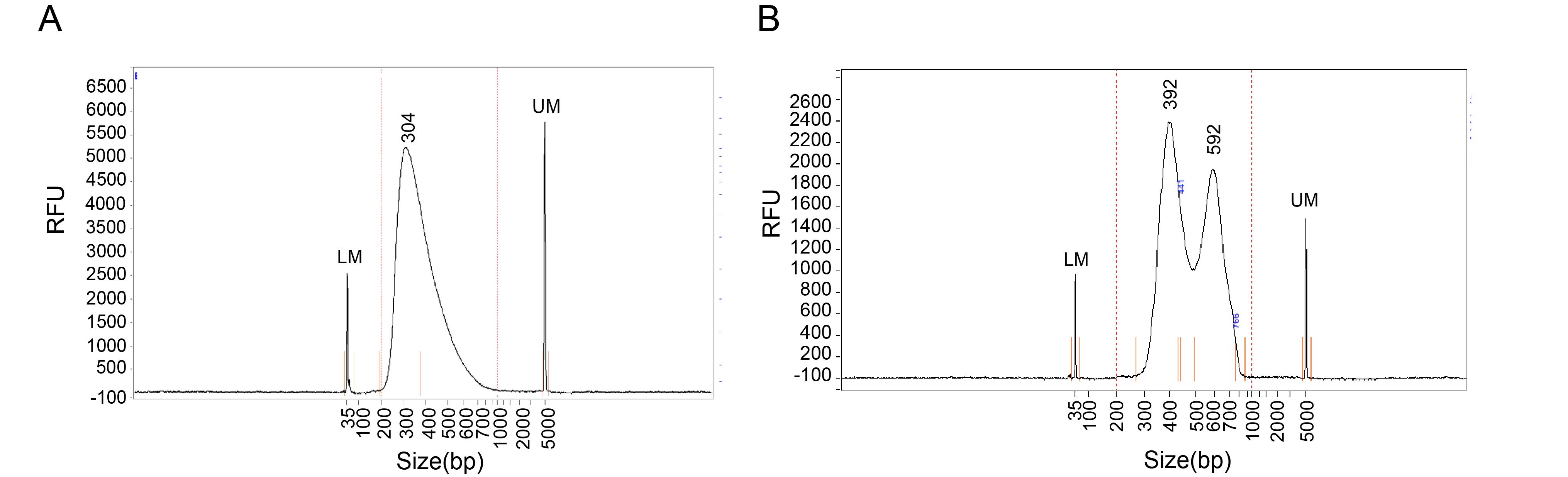
Figure 3. Fragment analyzer quality control of the cFOOT-seq, ATAC-cFOOT-seq, and cFOOT-ATAC-seq library. Representative fragment analyzer electropherogram showing the size distribution of a standard cFOOT-seq library (A) and cFOOT-ATAC-seq or ATAC-cFOOT-seq library (B). The x-axis indicates DNA fragment size (bp), and the y-axis indicates relative fluorescence units (RFU) of the library. The lower maker (LM) peak and upper marker (UM) peak define the sizing range, and the central peaks represent the main library fragments used for sequencing.
Troubleshooting
Problem 1: Poor cell integrity or cell loss.
Possible causes: Over-permeabilization, harsh lysis or washing conditions, excessive mechanical stress, or high centrifugation speed.
Solutions:
a. Optimize permeabilization and lysis conditions by reducing detergent concentration and/or incubation time.
b. Minimize mechanical stress during resuspension and mixing.
c. Use moderate centrifugation speeds to avoid cell or nuclear damage.
d. To reduce cell loss during supernatant removal, pellet samples by centrifugation in alternating orientations when possible, and remove supernatant in two steps: first, aspirate the bulk volume using a large-bore pipette tip, then briefly re-spin and remove the remaining small volume with a fine pipette tip.
e. Verify cell counts before and after key processing steps and standardize handling procedures across samples.
Problem 2: Low genomic DNA yield after extraction.
Possible causes:
a. Low effective input due to poor cell integrity or cell loss during sample preparation (see Problem 1).
b. Loss of DNA pellet during precipitation or wash steps.
c. Over-drying of the DNA pellet, resulting in incomplete DNA re-solubilization.
Solutions:
a. If low DNA yield is accompanied by cell fragmentation or loss, refer to Problem 1 and optimize cell handling and permeabilization conditions.
b. During DNA precipitation and washing, carefully retain the DNA pellet and avoid aspirating the pellet together with the supernatant.
c. Avoid over-drying the DNA pellet. If the pellet is over-dried, extend resuspension time and gently mix to ensure complete DNA re-solubilization. Confirm DNA recovery before proceeding to library construction.
Problem 3: Abnormal library fragment size distribution or loss of ATAC laddering.
Observed outcomes: cFOOT-seq libraries lack enrichment around ~300–500 bp; ATAC-integrated libraries lack characteristic nucleosomal laddering.
Possible causes:
a. For cFOOT-seq, inefficient or uneven sonication result in insufficient DNA fragmentation.
b. For ATAC-integrated workflows, suboptimal ATAC tagmentation conditions.
c. For cFOOT-ATAC, excessively high cytosine conversion rates can disrupt DNA duplex structure and reduce tagmentation efficiency, leading to poor ATAC enrichment.
Solutions:
a. For cFOOT-seq, optimize sonication conditions to achieve a predominant fragment size around ~300–500 bp.
b. For ATAC-integrated workflows, re-optimize tagmentation conditions (e.g., enzyme amount and reaction time) to restore nucleosome laddering.
c. For cFOOT-ATAC, adjust deamination conditions to maintain a moderate genome-wide conversion rate (typically ~30–35%), which is compatible with efficient ATAC enrichment.
Problem 4: Genome-wide cytosine conversion rate does not fall within the target range.
Possible causes: Suboptimal SsdAtox input, inappropriate deamination time, or limited enzyme access due to insufficient nuclear permeabilization.
Solutions: Perform SsdAtox titration using equal numbers of cells and select the enzyme input that yields the target conversion range (25%–35% for cFOOT-seq and cFOOT-ATAC-seq; 35%–45% for ATAC-cFOOT-seq). If the target conversion range cannot be achieved by adjusting enzyme input alone, then fine-tune deamination time and optimize nuclear permeabilization conditions as needed.
Problem 5: Weak or non-reproducible TF footprints.
Possible causes:
a. Low genome-wide conversion efficiency (often problematic when <25%).
b. Insufficient sequencing depth/usable converted reads.
c. Batch/handling variability, especially during deamination (enzyme amount, temperature, timing).
Solutions:
a. Ensure genome-wide conversion efficiency is ≥25% and keep it consistent across compared samples.
b. Increase sequencing depth (focus on usable converted reads).
c. Standardize the deamination step and compare within the same batch when possible.
Acknowledgments
Method development and optimization, M.-C.Y., H.W., and A.W.; Supervision, J.-M.Z., S.G., J.S., and X.L.; Writing—original draft, M.-C.Y.; Graphic design and schematic illustrations, H.W.; Data analysis and computational framework, A.W.; Writing—review and editing, J.-M.Z.
This work was supported by the National Key Research and Development Program of China (2021YFA1102000, 2022YFA1106000, and 2021YFA1102900), the National Natural Science Foundation of China (32270775, 81902583, 32270702, 32370868, and 32070802), the Fundamental Research Funds for the Central Universities (22120250374), and the Peak Disciplines (Type IV) of Institutions of Higher Learning in Shanghai.
This protocol is based on our previously published work [24], in which the cFOOT-seq method was originally described and validated.
Competing interests
The authors declare that they have filed an international patent application and a national patent application in China related to the cFOOT-seq method described in this manuscript.
References
- Lambert, S. A., Jolma, A., Campitelli, L. F., Das, P. K., Yin, Y., Albu, M., Chen, X., Taipale, J., Hughes, T. R. and Weirauch, M. T. (2018). The Human Transcription Factors. Cell. 172(4): 650–665. https://doi.org/10.1016/j.cell.2018.01.029
- Kim, S. and Wysocka, J. (2023). Deciphering the multi-scale, quantitative cis-regulatory code. Mol Cell. 83(3): 373–392. https://doi.org/10.1016/j.molcel.2022.12.032
- de Boer, C. G. and Taipale, J. (2024). Hold out the genome: a roadmap to solving the cis-regulatory code. Nature. 625(7993): 41–50. https://doi.org/10.1038/s41586-023-06661-w
- Johnson, D. S., Mortazavi, A., Myers, R. M. and Wold, B. (2007). Genome-wide mapping of in vivo protein-DNA interactions. Science. 316(5830): 1497–1502. https://doi.org/10.1126/science.1141319
- Skene, P. J. and Henikoff, S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. eLife. 6: e21856. https://doi.org/10.7554/eLife.21856
- Kaya-Okur, H. S., Wu, S. J., Codomo, C. A., Pledger, E. S., Bryson, T. D., Henikoff, J. G., Ahmad, K. and Henikoff, S. (2019). CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun. 10(1): 1930. https://doi.org/10.1038/s41467-019-09982-5
- Carter, B., Ku, W. L., Kang, J. Y., Hu, G., Perrie, J., Tang, Q. and Zhao, K. (2019). Mapping histone modifications in low cell number and single cells using antibody-guided chromatin tagmentation (ACT-seq). Nat Commun. 10(1): 3747. https://doi.org/10.1038/s41467-019-11559-1
- Wang, Q., Xiong, H., Ai, S., Yu, X., Liu, Y., Zhang, J. and He, A. (2019). CoBATCH for High-Throughput Single-Cell Epigenomic Profiling. Mol Cell. 76(1): 206–216 e207. https://doi.org/10.1016/j.molcel.2019.07.015
- Harada, A., Maehara, K., Handa, T., Arimura, Y., Nogami, J., Hayashi-Takanaka, Y., Shirahige, K., Kurumizaka, H., Kimura, H. and Ohkawa, Y. (2019). A chromatin integration labelling method enables epigenomic profiling with lower input. Nat Cell Biol. 21(2): 287–296. https://doi.org/10.1038/s41556-018-0248-3
- Altemose, N., Maslan, A., Smith, O. K., Sundararajan, K., Brown, R. R., Mishra, R., Detweiler, A. M., Neff, N., Miga, K. H., Straight, A. F., et al. (2022). DiMeLo-seq: a long-read, single-molecule method for mapping protein-DNA interactions genome wide. Nat Methods. 19(6): 711–723. https://doi.org/10.1038/s41592-022-01475-6
- Yue, X., Xie, Z., Li, M., Wang, K., Li, X., Zhang, X., Yan, J. and Yin, Y. (2022). Simultaneous profiling of histone modifications and DNA methylation via nanopore sequencing. Nat Commun. 13(1): 7939. https://doi.org/10.1038/s41467-022-35650-2
- Hesselberth, J. R., Chen, X., Zhang, Z., Sabo, P. J., Sandstrom, R., Reynolds, A. P., Thurman, R. E., Neph, S., Kuehn, M. S., Noble, W. S., et al. (2009). Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 6(4): 283-289. https://doi.org/10.1038/nmeth.1313
- Vierstra, J., Lazar, J., Sandstrom, R., Halow, J., Lee, K., Bates, D., Diegel, M., Dunn, D., Neri, F., Haugen, E., et al. (2020). Global reference mapping of human transcription factor footprints. Nature. 583(7818): 729–736. https://doi.org/10.1038/s41586-020-2528-x
- Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. and Greenleaf, W. J. (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 10(12): 1213–1218. https://doi.org/10.1038/nmeth.2688
- Hu, Y., Horlbeck, M. A., Zhang, R., Ma, S., Shrestha, R., Kartha, V. K., Duarte, F. M., Hock, C., Savage, R. E., Labade, A., et al. (2025). Multiscale footprints reveal the organization of cis-regulatory elements. Nature. 638(8051): 779–786. https://doi.org/10.1038/s41586-024-08443-4
- He, H. H., Meyer, C. A., Hu, S. S., Chen, M. W., Zang, C., Liu, Y., Rao, P. K., Fei, T., Xu, H., Long, H., et al. (2014). Refined DNase-seq protocol and data analysis reveals intrinsic bias in transcription factor footprint identification. Nat Methods. 11(1): 73–78. https://doi.org/10.1038/nmeth.2762
- Li, Z., Schulz, M. H., Look, T., Begemann, M., Zenke, M. and Costa, I. G. (2019). Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20(1): 45. https://doi.org/10.1186/s13059-019-1642-2
- Bentsen, M., Goymann, P., Schultheis, H., Klee, K., Petrova, A., Wiegandt, R., Fust, A., Preussner, J., Kuenne, C., Braun, T., et al. (2020). ATAC-seq footprinting unravels kinetics of transcription factor binding during zygotic genome activation. Nat Commun. 11(1): 4267. https://doi.org/10.1038/s41467-020-18035-1
- Kelly, T. K., Liu, Y., Lay, F. D., Liang, G., Berman, B. P. and Jones, P. A. (2012). Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 22(12): 2497–2506. https://doi.org/10.1101/gr.143008.112
- Krebs, A. R., Imanci, D., Hoerner, L., Gaidatzis, D., Burger, L. and Schubeler, D. (2017). Genome-wide Single-Molecule Footprinting Reveals High RNA Polymerase II Turnover at Paused Promoters. Mol Cell. 67(3): 411–422 e414. https://doi.org/10.1016/j.molcel.2017.06.027
- Shipony, Z., Marinov, G. K., Swaffer, M. P., Sinnott-Armstrong, N. A., Skotheim, J. M., Kundaje, A. and Greenleaf, W. J. (2020). Long-range single-molecule mapping of chromatin accessibility in eukaryotes. Nat Methods. 17(3): 319–327. https://doi.org/10.1038/s41592-019-0730-2
- Stergachis, A. B., Debo, B. M., Haugen, E., Churchman, L. S. and Stamatoyannopoulos, J. A. (2020). Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science. 368(6498): 1449–1454. https://doi.org/10.1126/science.aaz1646
- He, R., Dong, W., Wang, Z., Xie, C., Gao, L., Ma, W., Shen, K., Li, D., Pang, Y., Jian, F., et al. (2024). Genome-wide single-cell and single-molecule footprinting of transcription factors with deaminase. Proc Natl Acad Sci USA. 121(52): e2423270121. https://doi.org/10.1073/pnas.2423270121
- Wang, H., Wu, A., Yang, M. C., Zhou, D., Chen, X., Shi, Z., Zhang, Y., Liu, Y. X., Chen, K., Wang, X., et al. (2025). Genome-wide investigation of transcription factor footprints and dynamics using cFOOT-seq. Protein Cell. 16(11): 932–952. https://doi.org/10.1093/procel/pwaf071
- Roh, H., Shen, S. P., Hu, Y., Kwok, H. S., Siegenfeld, A. P., Lee, C., Zepeda, M. U., Guo, C. J., Roseman, S. A., Comenho, C., et al. (2025). Coupling CRISPR scanning with targeted chromatin accessibility profiling using a double-stranded DNA deaminase. Nat Methods. 22(10): 2083–2093. https://doi.org/10.1038/s41592-025-02811-2
- Swanson, E. G., Mao, Y., Mallory, B. J., Vollger, M. R., Bohaczuk, S. C., Oliveira, C. B., Lyon, D. B., Ranchalis, J., Parmalee, N. L., Cohen, B. A., et al. (2025). Mapping single-cell diploid chromatin fiber architectures using DAF-seq. Nat Biotechnol.: e1038/s41587–025–02914–3. https://doi.org/10.1038/s41587-025-02914-3
- Yu, T., Li, Z., Gibbs, E., Iwase, R., Francoeur, M. J., Phan, Q. V., Zhao, J., Rosin, J., Cole, P. A., Pinello, L., et al. (2024). Deaminase-mediated chromatin accessibility profiling with single-allele resolution. bioRxiv. https://doi.org/10.1101/2024.12.17.628768
- de Moraes, M. H., Hsu, F., Huang, D., Bosch, D. E., Zeng, J., Radey, M. C., Simon, N., Ledvina, H. E., Frick, J. P., Wiggins, P. A., et al. (2021). An interbacterial DNA deaminase toxin directly mutagenizes surviving target populations. eLife. 10: E62967. https://doi.org/10.7554/eLife.62967
- Consortium, I. T. P.-C. A. o. W. G. (2020). Pan-cancer analysis of whole genomes. Nature. 578(7793): 82–93. https://doi.org/10.1038/s41586-020-1969-6
Article Information
Publication history
Received: Dec 23, 2025
Accepted: Feb 11, 2026
Available online: Feb 27, 2026
Published: Mar 20, 2026
Copyright
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Yang, M. C., Wu, A., Wang, H., Liu, X., Shi, J., Gao, S. and Zhang, J. M. (2026). A Cytosine Deaminase–Based Genomic Footprinting Assay (cFOOT-seq) for Detecting Transcription Factor Occupancy. Bio-protocol 16(6): e5637. DOI: 10.21769/BioProtoc.5637.
Category
Developmental Biology > Cell signaling
Molecular Biology > DNA > DNA-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.