- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Identification of Topologically Associating Domains (TADs) and Long-distance Enhancer–gene/gene–gene Interactions with Hi-C and HiChIP

Published: Nov 20, 2023 DOI: 10.21769/BioProtoc.4879 Views: 502
Reviewed by: Anonymous reviewer(s)

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The hierarchy featured architecture of 3D genome influences the expression of genes to regulate development and defense processes. Hi-C is one of the most widespread technologies to explore hierarchy 3D genome organizations, including compartment, topologically associating domains (TADs), and loops. HiChIP can accurately identify chromatin loops between cis-regulatory elements in non-coding regions and genes. At the same time, growing innovative methods can be used to identify different hierarchy architectures of 3D genome, but most analyses are performed with a combination of a variety of software. Therefore, a clear and complete pipeline is essential. We summarize the detailed usage of various tools for identifying the 3D genome structure using Hi-C and HiChIP, as well as for visualizing the final result. Using these software tools, we identify the TADs and loops from the Hi-C and HiChIP libraries in human cell lines and cotton plants. In brief, this pipeline will help researchers choose a suitable tool with less time cost.
Keywords: 3D genomeBackground
The genetic diversity of organisms is the fundamental basis for forming complex and diverse global ecosystems. Adequate and comprehensive knowledge of genetic information will help us understand the nature of evolution more deeply and thus improve our living environment without affecting the survival of other species. The study of the interpretation of genetic information from multiple perspectives has been booming (Misteli, 2020; Purugganan and Jackson, 2021; Song et al., 2021; Zhang et al., 2021). Among them, the 3D genome has become a fascinating study field. It has been extensively used to comprehensively study how the hierarchy of 3D genome regulates biological processes such as evolution, development, and resistance to pathogens by combining 1D genomics, transcriptomics, and epigenomics (Fullwood et al., 2009; Lieberman-Aiden et al., 2009; Mumbach et al., 2017; Sun et al., 2018; Wang et al., 2018; Norrie et al., 2019; Zheng and Xie, 2019; Yang et al., 2022). So far, there is a growing body of literature on topologically associating domains (TADs) and loops, since they are finely architected to regulate changes in genetic information (Rao et al., 2014; Wang et al., 2018; Zheng et al., 2019; Espinola et al., 2021; Hoencamp, 2021). Hence, accurate identification of TAD and loop structures is essential for understanding how the hierarchy of 3D genome architecture regulates biological processes.
With the innovation in experimental methods of 3D genomic study and the advancement of sequencing technologies, a large number of software tools were developed to identify 3D genome structures (Ay et al., 2014; Wang et al., 2015; Forcato et al., 2017; Bhattacharyya et al., 2019; Yardımcı et al., 2019; Fernandez et al., 2020; Wolff et al., 2020; Mourad, 2022). They have contributed to a boom in the field of 3D genomic research. This also increases the difficulties for researchers to realize them (Forcato et al., 2017). Therefore, a complete order pipeline is very useful for scientific researchers. To achieve this, we summarized the processing of several software tools, including identifying TADs by TADLib and Juicer, inferring loops of Hi-C data by Fit-Hi-C and Juicer, and inferring loops of HiChIP data by FitHiChIP and hichipper. The integration of these software into a whole pipeline by combining HiC-Pro and Juicebox can provide final visualization results from raw reads (Figure 1). The pipeline provides a comprehensive 3D genome analysis process that is applicable not only to Hi-C datasets but also compatible with HiChIP datasets. Moreover, the pipeline eliminates the need for complex intermediate steps in multiple software, providing users a simple and efficient experience while preserving the software's customizable function. In summary, the pipeline provides user-friendly features that are especially beneficial for researchers who are new to 3D genome analysis.
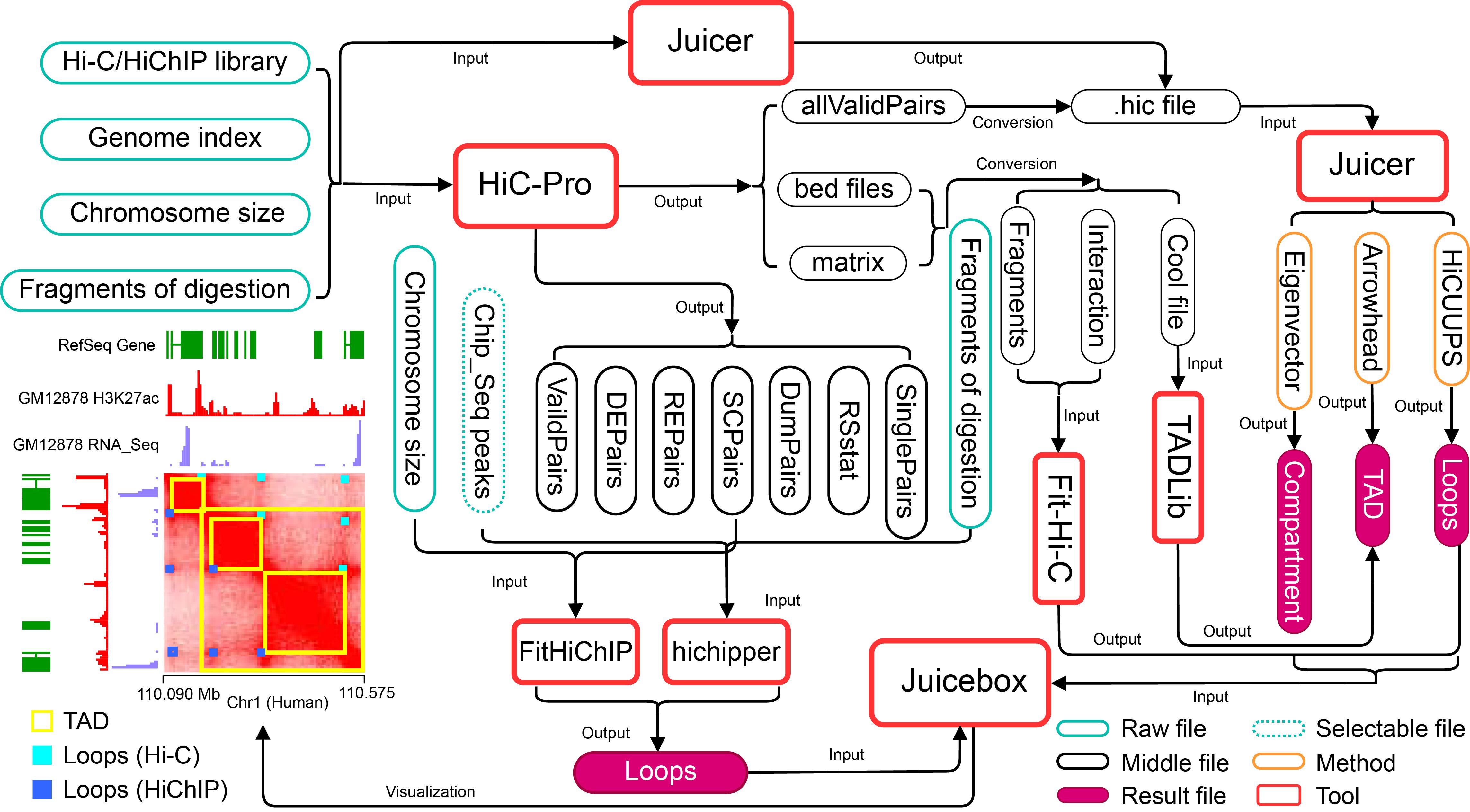
Figure 1. Flowchart for the identification of topologically associating domains (TAD) and loops from the datasets of Hi-C and HiChIP by different software
Software and datasets
Software
Python (Version 3.9.5/Version 2.7.18, https://www.python.org/downloads/) (2020/09)
R (Version 4.0.0, https://cran.r-project.org/bin/windows/base/old/) (2020/09)
Bowtie2 (Version 2.4.4) (Langmead and Salzberg, 2012)
BWA (Version 0.7.17) (Li and Durbin, 2009)
SAMtools (Version 1.9) (Li et al., 2009)
BEDTools (Version 2.27) (Quinlan and Hall, 2010)
MACS2 (Version 2.1.1) (Zhang et al., 2008)
HiC-Pro (Version 2.11.4) (Servant et al., 2015)
Juicer (Version 1.6) (Durand et al., 2016)
Juicer tools jar (Version 1.22.01, https://github.com/aidenlab/juicer/wiki/Download) (2020/11)
Juicebox (Version 1.11.08, https://github.com/aidenlab/Juicebox/wiki/Download) (2020/11)
TADLib (Version 0.4.1) (Wang et al., 2015)
HiCPeaks (Version 0.3.4, https://github.com/XiaoTaoWang/HiCPeaks) (2021/04)
Fit-Hi-C (Version 2.0.8) (Ay et al., 2014)
FitHiChIP (Version 9.1) (Bhattacharyya et al., 2019)
hichipper (Version 0.7.7) (Lareau and Aryee, 2018)
Data
Library of Hi-C
Hi-C data of human GM12878 B-lymphoblastoid cells (Rao et al., 2014)
Hi-C data of cotton fiber of Gossypium barbadense 3–79 at 20 days post anthesis (DPA) (Pei et al., 2022)
Library of HiChIP
HiChIP data (H3K27ac) of human GM12878 B-lymphoblastoid cells (Mumbach et al., 2017)
Procedure
HiC-Pro (Servant et al., 2015)
The HiC-Pro is a pipeline for Hi-C data processing, from a raw Hi-C dataset to a normalized contact matrix by reads aligning, reads pairing, pairs dumping, and contact map building. The bowtie2 index, the restriction fragments after digestion, and chromosome size information are necessary files for processing HiC-Pro. Raw interaction matrix is used for subsequent analysis.
Construct bowtie2 index that will be used to reads alignment by bowtie2.
$ bowtie2-build reference_genome.fa prefix_of_output_file
Calculate the chromosome size that is needed to build Hi-C matrix.
$ samtools faidx reference_genome.fa
$ awk -v OFS = “\t” ‘{print $1, $2}’ reference_genome.fa.fai > chromosome_size.bed
Create enzyme fragment file to get Hi-C fragment information.
$ python HiC-Pro/bin/utils/digest_genome.py reference_genome.fa -r mboi/dpnii/bglii/hindiii -o output_file_name (python 2.7.18)
Rebuild the config file of HiC-Pro that adds the path of bowtie2 index, chromosome size, and fragment of enzymatic digestion.
$ BOWTIE2_IDX_PATH = “The path of bowtie2 index produced in step 1”
$ GENOME_SIZE = “The file of single chromosome size produced in step 2”
$ GENOME_FRAGMENT = “The file of fragments produced in step 3”
Run HiC-Pro on a personal computer in a standalone model.
$ HiC-Pro -i path_input_file (raw reads) -o path_output_file -c you_config_file
Run HiC-Pro on a cluster.
$ HiC-Pro -i path_input_file (raw reads) -o path_output_file_path -c you_config_file -p
$ bsub < HiCPro_step1.sh # Parallel workflow
$ bsub < HiCPro_step2.sh # Merge all outputs in single thread
Juicer (Durand et al., 2016)
Juicer is a platform that integrates producing Hi-C matrix from Hi-C library, identifying compartment (Eigenvector), presuming TAD (Arrowhead), and inferring loops (HiCCUPS). Juicer aligns reads to the reference genome using BWA and requires an enzyme cut site file.
Construct BWA index files.
$ bwa index reference_genome.fa -p prefix_of_output_file
Calculate the size of a single chromosome.
$ samtools faidx reference_genome.fa
$ awk -v OFS = “\t” ‘{print $1, $2}’ reference_genome.fa.fai > chromosome_size.bed
Generate an enzyme cut sites file. The enzymes available include HindIII, DpnII, MobI, Sau3AI, and Arima. Also, you can add other enzyme cut fragments in the python file.
$ python juicer/misc/generate_site_positions.py DpnII prefix_of_output_file reference_genome.fa (python 3.9.5)
The “rawdata,” “references,” “restriction_sites,” and “scripts” folders should preferably exist in the working directory. The rawdata folder includes the “fastq” folder where the Hi-C library is stored. The references folder includes bwa index, genome, and chromosome size. The enzyme cut sites file is stored in the restriction_sites folder. The scripts folder includes all scripts that need to be run. Copy the juicer/cpu/common folder into the scripts folder.
$ juicer/cpu/juicer.sh -d full_path_of_fastq_file -z reference_genome.fa -g genome_name -D workdir -p chromosome_size -y full_path_of_enzyme_cut_site -t number_of_threads
Identify TAD by Juicer with arrowhead algorithm. Users should download juicer_tools.jar and put it in the scripts folder.
$ java -jar scripts/juicer_tools.jar arrowhead -m size_of_the_sliding_window (must be an even number) -r resolution -k normalization_method (NONE/VC/VC_SQRT/KR) --threads number_of_threads *.hic_file directory_of_output
Medium resolution maps: -m 2,000 -r 10,000 -k KR. High resolution maps: -m 2,000 -r 5,000 -k KR.
Infer loops from a .hic file data by using HiCCUPS algorithm.
$ java -jar scripts/juicer_tools.jar hiccups --cpu -r resolution -f values_of_FDR -p width_of_peak -i width_of_window -d distances_used_for_merging_nearby_pixels -t thresholds_for_merging_loop -k normalizations_methods (NONE/VC/VC_SQRT/KR) --threads number_of_threads *.hic_file directory_of_output
Convert the result of HiC-Pro to *.hic file.
$ java -Xmx2g -jar juicebox_tools.jar pre -r resolution *.VaildPairs *.hic genome_name
The memory can be expanded by adjusting the parameter -Xmx2g to a desired value, such as -Xmx500g,whicht can allocate 500 gigabytes of memory.
TADLib (Wang et al., 2015)
TADLib uses a machine-learning approach to predict TAD structure after training the model. The contact matrix produced by HiC-Pro is used as an input file of TADLib after conversion to a cool file format.
Convert result files of HiC-Pro to the input file of TADLib. The intrachromosomal interactions are used to identify TAD structures by TADLib, so they must be selected.
Choose single chromosome interaction matrix.
$ awk -v OFS=“\t” ‘{if(($1>=chr_bin_start)&&($1<=chr_bin_end)&&($2>=chr_bin_start)&&($2<=chr_bin_end)) print $1-chr_bin_start+1,$2-chr_bin_start+1,$3}’ matrix_file > result_file (The format of file name: ChromosomeNumber_ChromosomeNumber.txt)
Merge all single chromosome interaction matrices.
$ toCooler -O output_filename.cool -d meta_file --chromsizes-file reference_ChrSize.bed --no-banlance
The format of meta file.
res: The size of resolution
rep1: path_of_single_chromosome_interaction_matrix_of_repeat1
rep2: path_of_single_chromosome_interaction_matrix_of_repeat2
Identify TAD by TADLib.
$ hitad -O output_TAD_filename.bed -d meta_file --logFile hitad.log -p number_of_threads -W RAW --maxsize Largest_TAD_size
The format of meta file.
res: The size of resolution
rep1: cool_file::The size of resolution
rep2: cool_file::The size of resolution
Fit-Hi-C (Ay et al., 2014)
Fit-Hi-C can infer significant interaction loops by balancing distance, strength, and probability of two interaction loci. The input file of Fit-Hi-C from the interaction matrix is produced by HiC-Pro.
Transform of HiC-Pro result file to Fit-Hi-C input file.
$ python Fithic/hicpro2fithic.py -i filename.matrix -b filename_abs.bed -r resolution_of -o output_file_directory -n prefix_of_output_file (python 3.9.5)
Infer loops by Fit-Hi-C.
$ fithic -f fragment -i interaction -r resolution -L lowerbound -U upperbound -p number_of_spline_passes -o output_file_directory -l prefix_of_output_file
FitHiChIP (Bhattacharyya et al., 2019)
FitHiChIP infers significant cis interactions from a given HiChIP/PLAC-seq experiment. In addition to calling peaks of histone modification sites from the HiChIP library, it can also combine existing peak data. For calling peaks from the HiChIP library, *.ValidPairs, *.DEPairs, *.REPairs, and *.SCPairs files generated by HiC-Pro are required. At the same time, the available pairwise interaction file generated by HiC-Pro is required as the input file of FitHiChIP. In addition, chromosome size information is also needed.
Run HiC-Pro to produce the available pairwise interaction file.
Call peaks in the HiChIP library by FitHiChIP.
$ sh FitHiChIP/Imp_Scripts/PeakInferHiChIP.sh -H Path_of_HiC-Pro -D Path_of_output -R Chromosome_size -M Parameters_of_macs2
Path of HiC-Pro include *.ValidPairs, *.DEPairs, *.REPairs, *.SCPairs and *.ValidPairs files.
Modify the config file of FitHiChIP.
$ ValidPairs = The valid pairs result file produced by HiC-Pro
$ PeakFile = File containing reference ChIP-seq/HiChIP peaks
$ OutDir = Path of the result
$ ChrSizeFile = Chromosome size files for reference genomes
$ BINSIZE = size of the bins
$ QVALUE = FDR value
$ UseP2Pbackgrnd = Two possible values 0 or 1. 0 represent loose background, 1 represent stringent background.
$ BiasType = Can be 1 (coverage bias) or 2 (ICE bias).
Run FitHiChIP.
$ bash FitHiChIP_HiCPro.sh -C config_file
hichipper (Lareau and Aryee, 2018)
The hichipper package can be used to determine library quality, identify and characterize DNA loops, and interactively visualize loops. This package takes the output from a HiC-Pro run. Similar to FitHiChIP, hichipper can also be used to call peaks from the HiChIP library or select an existing peak file.
Run HiC-Pro to produce some input files of hichipper.
Modify the configuration file that call peaks of histone modification sites in the HiChIP library by hichipper. Configuration parameters are written in the .yaml file. hichipper aggregates reads density from either all samples (COMBINED) or each sample (EACH) individually. Additionally, users can specify whether all reads density (ALL) is used or whether only self-ligation reads are used (SELF). The result path of HiC-Pro includes *.DEPairs, *.DumpPairs, *.RSstat, *.SCPairs, *.SinglePairs, and *.ValidPairs files.
The format of config.
peaks:
- COMBINED/EACH, ALL/ SELF
resfrags:
- The file of the enzyme section
hicpro_output:
- The path of the HiC-Pro output file
Modify the config file to identify loops by hichipper.
Peaks:
- The file of peaks
resfrags:
- The file of the enzyme section
hicpro_output:
- The path of the HiC-Pro output file
Run hichipper to identify loops of HiChIP data.
$ hichipper –out path_output_file config_file
Data analysis
Result interpretation
By using the above parameters of Juicer, TADs and loops were identified from the GM12878 library. The result is generally consistent with previous studies (Figure 2A). TADLib and Fit-Hi-C were used to identify TADs and loops in cotton. The results show that the loop interaction primarily occurs in TAD interior (Figure 2B). A comparison of the loops inferred by FitHiChIP and hichipper shows that most of them are the same, and a few differences may be due to different algorithms (Figure 2C).
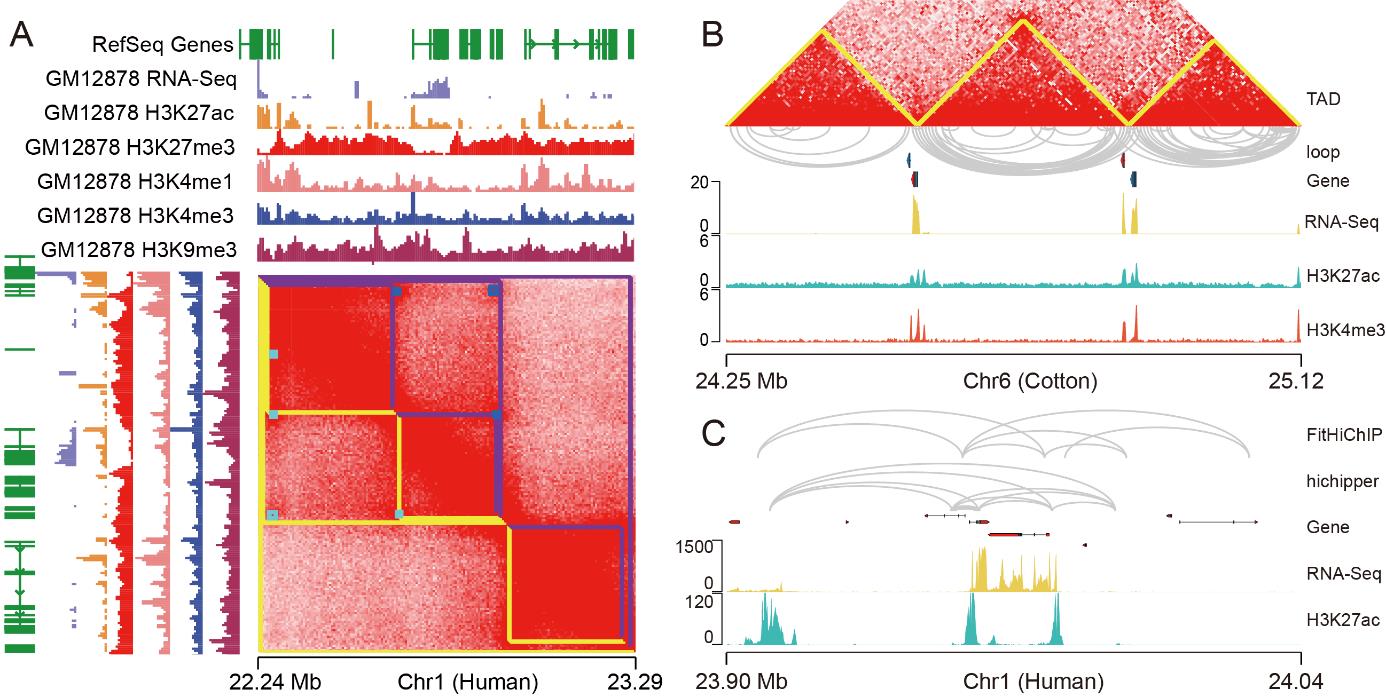
Figure 2. Topologically associating domains (TAD) and loops were identified from Hi-C and HiChIP library by different software. A. The TAD and loop were identified by Juicer in GM12878 Hi-C library. The purple line and the blue point on the heat map represent TADs and loops identified in a previous study (Durand et al., 2016). The yellow and cyan lines on the heat map represent TADs and loops identified by Juicer. B. The TAD and loops were identified by TADLib and Fit-Hi-C in Gossypium barbadense 3-79 library. C. The loops of GM12878 HiChIP (H3K27ac) library were inferred by FitHiChIP and hichipper. The data of ChIP-Seq with different antibodies and RNA-Seq were from previous studies (Rao et al., 2014; Davis et al., 2018; Pei et al., 2022).
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2021YFF1000100) and the National Natural Science Foundation of China (32170645, 31922069). Appreciation to all research scholars who participated in the software of pipeline development and provided experimental data.
Competing interests
The authors declare no competing interests.
References
- Ay, F., Bailey, T. L. and Noble, W. S. (2014). Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res. 24(6): 999-1011.
- Bhattacharyya, S., Chandra, V., Vijayanand, P. and Ay, F. (2019). Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nat. Commun. 10(1): 4221.
- Davis, C. A., Hitz, B. C., Sloan, C. A., Chan, E. T., Davidson, J. M., Gabdank, I., Hilton, J. A., Jain, K., Baymuradov, U. K., Narayanan, A. K. et al. (2018). The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic. Acids. Res. 46(D1): D794-D801.
- Durand, N. C., Shamim, M. S., Machol, I., Rao, S. S., Huntley, M. H., Lander, E. S. and Aiden, E. L. (2016). Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 3(1): 95-98.
- Espinola, S. M., Gotz, M., Bellec, M., Messina, O., Fiche, J. B., Houbron, C., Dejean, M., Reim, I., Cardozo Gizzi, A. M., Lagha, M. and Nollmann, M. (2021). Cis-regulatory chromatin loops arise before TADs and gene activation, and are independent of cell fate during early Drosophila development. Nat. Genet. 53(4): 477-486.
- Fernandez, L. R., Gilgenast, T. G. and Phillips-Cremins, J. E. (2020). 3DeFDR: statistical methods for identifying cell type-specific looping interactions in 5C and Hi-C data. Genome Biol .21(1): 219.
- Forcato, M., Nicoletti, C., Pal, K., Livi, C. M., Ferrari, F. and Bicciato, S. (2017). Comparison of computational methods for Hi-C data analysis. Nature methods 14(7): 679.
- Fullwood, M. J., Liu, M. H., Pan, Y. F., Liu, J., Xu, H., Mohamed, Y. B., Orlov, Y. L., Velkov, S., Ho, A., Mei, P. H. et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462(7269): 58-64.
- Hoencamp, C. (2021). 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science 372(6545):984-989.
- Langmead, B. and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4): 357-359.
- Lareau, C. A. and Aryee, M. J. (2018). hichipper: a preprocessing pipeline for calling DNA loops from HiChIP data. Nat Methods 15(3): 155-156.
- Li, H. and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25(14): 1754-1760.
- Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., Marth, G., Abecasis, G., Durbin, R. and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25(16): 2078-2079.
- Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., Amit, I., Lajoie, B. R., Sabo, P. J., Dorschner, M. O. et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326(5950): 289-293.
- Misteli, T. (2020). The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 183(1): 28-45.
- Mourad, R. (2022). TADreg: a versatile regression framework for TAD identification, differential analysis and rearranged 3D genome prediction. BMC Bioinformatics 23(1): 82.
- Mumbach, M. R., Satpathy, A. T., Boyle, E. A., Dai, C., Gowen, B. G., Cho, S. W., Nguyen, M. L., Rubin, A. J., Granja, J. M., Kazane, K. R. et al. (2017). Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet. 49(11): 1602-1612.
- Norrie, J. L., Lupo, M. S., Xu, B., Al Diri, I., Valentine, M., Putnam, D., Griffiths, L., Zhang, J., Johnson, D., Easton, J. et al. (2019). Nucleome Dynamics during Retinal Development. Neuron 104(3): 512-528 e511.
- Pei, L., Huang, X., Liu, Z., Tian, X., You, J., Li, J., Fang, D. D., Lindsey, K., Zhu, L., Zhang, X. and Wang, M. (2022). Dynamic 3D genome architecture of cotton fiber reveals subgenome-coordinated chromatin topology for 4-staged single-cell differentiation. Genome Biol. 23(1): 45.
- Purugganan, M. D. and Jackson, S. A. (2021). Advancing crop genomics from lab to field. Nat Genet 53(5): 595-601.
- Quinlan, A. R. and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841-842.
- Rao, S. S., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., Sanborn, A. L., Machol, I., Omer, A. D., Lander, E. S. and Aiden, E. L. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7): 1665-1680.
- Servant, N., Varoquaux, N., Lajoie, B. R., Viara, E., Chen, C. J., Vert, J. P., Heard, E., Dekker, J. and Barillot, E. (2015). HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16: 259.
- Song, J. M., Xie, W. Z., Wang, S., Guo, Y. X., Koo, D. H., Kudrna, D., Gong, C., Huang, Y., Feng, J. W., Zhang, W. et al. (2021). Two gap-free reference genomes and a global view of the centromere architecture in rice. Mol. Plant. 14(10): 1757-1767.
- Sun, J. H., Zhou, L., Emerson, D. J., Phyo, S. A., Titus, K. R., Gong, W., Gilgenast, T. G., Beagan, J. A., Davidson, B. L., Tassone, F. and Phillips-Cremins, J. E. (2018). Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 175(1): 224-238 e215.
- Wang, M., Wang, P., Lin, M., Ye, Z., Li, G., Tu, L., Shen, C., Li, J., Yang, Q. and Zhang, X. (2018). Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nature Plants 4(2).
- Wang, Q., Sun, Q., Czajkowsky, D. M. and Shao, Z. (2018). Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun. 9(1): 188.
- Wang, X. T., Dong, P. F., Zhang, H. Y. and Peng, C. (2015). Structural heterogeneity and functional diversity of topologically associating domains in mammalian genomes. Nucleic Acids. Re 43(15): 7237-7246.
- Wolff, J., Rabbani, L., Gilsbach, R., Richard, G., Manke, T., Backofen, R. and Gruning, B. A. (2020). Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids. Res. 48(W1): W177-W184.
- Yang, H., Zhang, H., Luan, Y., Liu, T., Yang, W., Roberts, K. G., Qian, M. X., Zhang, B., Yang, W., Perez-Andreu, V. et al. (2022). Noncoding genetic variation in GATA3 increases acute lymphoblastic leukemia risk through local and global changes in chromatin conformation. Nat. Genet. 54(2): 170-179.
- Yardımcı, G. G., Ozadam, H., Sauria, M. E. G., Ursu, O., Yan, K. K., Yang, T., Chakraborty, A., Kaul, A., Lajoie, B. R., Song, F., Zhan, Y. et al. (2019). Measuring the reproducibility and quality of Hi-C data. Genome Biol. 20(1).
- Zhang, K., Hocker, J. D., Miller, M., Hou, X., Chiou, J., Poirion, O. B., Qiu, Y., Li, Y. E., Gaulton, K. J., Wang, A., Preissl, S. and Ren, B. (2021). A single-cell atlas of chromatin accessibility in the human genome. Cell 184(24): 5985-6001 e5919.
- Zhang, Y., Liu, T., Meyer, C. A., Eeckhoute, J., Johnson, D. S., Bernstein, B. E., Nusbaum, C., Myers, R. M., Brown, M., Li, W. and Liu, X. S. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9(9): R137.
- Zheng, H. and Xie, W. (2019). The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell. Biol. 20(9): 535-550.
- Zheng, M., Tian, S. Z., Capurso, D., Kim, M., Maurya, R., Lee, B., Piecuch, E., Gong, L., Zhu, J. J., Li, Z. et al. (2019). Multiplex chromatin interactions with single-molecule precision. Nature 566(7745): 558-562.
Supplementary information
Data and code availability: All data and code have been deposited to GitHub: https://github.com/Bio-protocol/TAD-and-loop-identification-workflows.git.
Article Information
Copyright
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
How to cite
Huang, X., Zhu, L., Zhang, X. and Wang, M. (2023). Identification of Topologically Associating Domains (TADs) and Long-distance Enhancer–gene/gene–gene Interactions with Hi-C and HiChIP. Bio-101: e4879. DOI: 10.21769/BioProtoc.4879.
Category
Bioinformatics and Computational Biology
Systems Biology > 3D Genomics
Systems Biology > Genomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.