

Articles In Press
"Articles In Press"是经过同行评审并被接受发表的文章。在正式发表之前还可能有内容修改,但可以使用DOI对文章进行引用。正式发表后,该文章将不再在此处展示,现有链接将自动重定向到文章的最终版本。
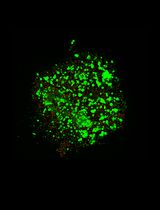
Generation and Characterization of Adaptive Anoikis-Resistant Cells Using Cyclic Attachment-Detachment Culture of Cancer Cells
Anoikis resistance, or the ability of cancer cells to evade cell death triggered by immediate detachment from the extracellular matrix, is a critical established hallmark of metastatic cancer. While suspension culture models have been used to study anoikis, most focus on defined single time points or prolonged suspension that may not recapitulate the effects of repeated stress that tumor cells experience during metastatic dissemination. Here, we describe a detailed protocol for generating anoikis-resistant (AnR) cancer cells that have adapted to such stress through exposure to repeated cycles of suspension stress on poly-HEMA-coated plates, followed by recovery under standard attached conditions. The protocol includes methods for determining baseline anoikis sensitivity, generating AnR cells over 7–9 attachment-detachment cycles, assessing the stability and reversion of the anoikis-resistant phenotype, and characterizing AnR cells using Live/Dead staining of spheroids, flow cytometry–based apoptosis assays, and immunofluorescence for proliferation markers. This approach produces a non-genetic, reversible anoikis-resistant state that models the adaptive transcriptional reprogramming underlying metastatic progression, providing a reproducible and physiologically relevant in vitro system for studying anoikis resistance mechanisms and evaluating therapeutic strategies for prevention and reversal of such adaptations.
A Dual-gRNA CRISPR/Cas9 System for Efficient Generation of Large Fragment Deletions in Poplar
CRISPR/Cas9-based genome editing is a powerful approach for functional genomics and bioenergy research in woody plants. However, conventional single guide RNA (gRNA) strategies predominantly generate small insertions or deletions that may not fully disrupt gene function and often require extensive sequencing for mutation identification. Here, we present an optimized protocol for the efficient generation of large-fragment deletion mutants in Populus tremula × P. alba clone INRA 717-1B4 using a dual-gRNA CRISPR/Cas9 system. Co-expression of two gRNAs flanking the target region induces double-strand breaks at both sites, enabling the deletion of the intervening genomic fragment, typically larger than 50 bp. This protocol describes step-by-step procedures for gRNA design, vector construction, Agrobacterium-mediated transformation, plant regeneration, and molecular validation. Using the PtFBX230 gene as a representative target, large deletions are readily identified by conventional PCR and agarose gel electrophoresis, enabling rapid and cost-effective genotyping. This protocol can be readily adopted to other loci in poplar and related woody species and provides a robust framework for generating null alleles to support functional genomics and bioenergy-related trait engineering in woody plants.
Tracking AC Electric Stimulation–Induced Persistent Locomotion Behavior in the Nematode Caenorhabditis elegans
Persistent neural activity underlies fundamental brain functions such as memory, decision-making, and emotion. Despite its importance, experimental paradigms that enable quantitative analysis of persistent behavioral responses remain limited. Here, we describe a protocol to induce and measure a persistent locomotor response by applying a brief alternating current (AC) electric stimulus to the nematode Caenorhabditis elegans. This method reliably evokes a prolonged increase in locomotion speed that persists for minutes after stimulus termination and can be quantified by video tracking. Because C. elegans has a fully mapped connectome and is amenable to genetic and neurophysiological manipulation, this protocol provides a useful platform for dissecting the molecular and neural mechanisms underlying persistent behavioral responses. Electrically induced persistent locomotion serves as a simple, robust, and quantifiable behavioral readout for studying the regulation of neural persistence in vivo.
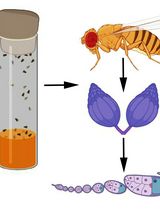
Visualizing Membrane Nanotube Dynamics in Drosophila Oocyte Using Live-Cell Imaging
Thin membrane protrusions in cells help them communicate, create traction forces during their movement, and coordinate complex development in multicellular organisms. These structures include cytonemes, tunneling nanotubes, and microtubule-based nanotubes (MT-nanotubes), each with a different cytoskeletal constitution and function. Actin-based cytonemes help deliver signaling molecules, while microtubule-based nanotubes assist with transporting vesicles and organelles. Despite their physiological role, we still do not fully understand how these thin membrane protrusions form and function. In this study, we introduce an improved live-cell imaging method to observe polar cell protrusions during micropyle morphogenesis in developing Drosophila eggs. This technique combines precise developmental staging, careful dissection, and optimized ex vivo culture conditions to maintain tissue health during extended imaging. We also fine-tuned the imaging settings to reduce phototoxicity and thermal stress. This allows for continuous, high-resolution tracking of protrusion dynamics in real time. Our protocol addresses major drawbacks of fixed-tissue methods by capturing the entire process of protrusion formation, extension, and remodeling in intact living tissue. Additionally, it works well with drugs, making it a useful tool for functional studies. Overall, this approach builds a strong foundation for exploring membrane protrusion biology. It can also be applied to investigate similar developmental processes in other systems, aiding our understanding of normal development and diseases.
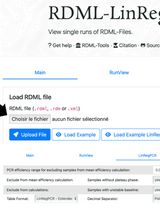
Efficiency-Corrected Relative Quantification of qPCR Data Using LinRegPCR and a Spreadsheet-Based Workflow
Quantitative real-time PCR (qPCR) is widely used for the quantitative assessment of relative transcript abundance in biological and medical research. Rigorous interpretation of qPCR data requires appropriate correction and normalization workflows that account for both technical variability and experimental heterogeneity. Regarding the correction step, the most used qPCR analysis relies on the 2-ΔΔCq method, which assumes identical and optimal amplification efficiencies across assays. Alternative strategies estimate amplification efficiencies using standard curves generated from serial dilutions, but these approaches require additional experimental work and may introduce serious dilution-related bias. Here, we describe a spreadsheet-based computational protocol for the correction of relative quantification of qPCR data that integrates amplification efficiencies derived directly from raw amplification curves using LinRegPCR. Cq values and per-reaction efficiency estimates are combined to calculate efficiency-corrected target quantities. Correction is then followed by normalization using the geometric mean of two reference genes. The workflow enables calculation of relative abundance fold-changes without the need for standard curves and produces output tables suitable for downstream statistical analysis. This protocol provides a transparent, dilution-free method for efficiency-corrected qPCR data analysis that can be implemented using commonly available software, facilitating reproducible and Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE)-compliant reporting of qPCR results.

A Flow Cytometry–Based Assay to Quantify the Binding of Transmembrane Ligands to Their Cognate Receptors Using Fluorescent Virus-Like Particles
The binding of transmembrane (TM) ligands to their cognate TM receptors on neighboring cells governs intercellular adhesion and direct cell–cell communication. However, these interactions are difficult to study in vitro because they depend on membrane presentation, ligand orientation, receptor clustering, and avidity, features often not captured by soluble recombinant ligands or cell-free assays. Here, we describe a flow cytometry–based assay using fluorescent, lentiviral virus-like particles (VLPs) displaying TM ligands to quantify binding to their receptors on target cells. Fluorescent VLPs are generated in-house by plasmid transfection in HEK293T cells and enable direct fluorescent detection without fluorochrome-conjugated secondary antibodies. The system is modular and readily accommodates engineered ligand constructs, including patient-derived variants. We applied this platform to generate ICAM-1-displaying fluorescent VLPs and to study human LFA-1 function in patient-derived leukocytes. This protocol provides a detailed workflow for VLP production and in vitro binding assays, offering a simple, quantitative, and cost-effective approach for studying TM ligand–receptor interactions in a membrane context. The system is well-suited for mechanistic studies, functional assessment of patient-derived variants, and direct binding assays using patient-derived cells. Integrating the assay into multicolor flow cytometry panels enables simultaneous immunophenotyping and quantification of up to four ligand–receptor interactions at single-cell resolution.

Stepwise Protocol for Alternative Splicing Analysis in Single-Cell SMART-Seq2 RNA-Seq Data
RNA alternative splicing (AS) is an essential process that expands transcriptomic and proteomic diversity in eukaryotic cells and contributes to cellular heterogeneity across physiological and pathological conditions in humans. With the advent of single-cell RNA sequencing (scRNA-seq), it has become possible to study AS at cellular resolution, although robust and standardized analytical workflows remain to be developed. Here, we present a stepwise protocol for analyzing AS in single cells from pediatric high-grade gliomas (pHGGs) harboring the histone H3.3 lysine 27-to-methionine (H3.3K27M) mutation using SMART-Seq2 scRNA-seq data. Starting from raw sequencing reads, the workflow includes read alignment, gene-level quantification, splice junction and intron quantification, and single-nucleotide variant-based mutation detection. Gene expression–based clustering and cell-type annotation are performed by using the Seurat R package. AS analysis in tumor cells is then conducted using the MARVEL R package in combination with customized scripts to calculate percent spliced-in (PSI) values, identify variable AS events, perform dimensionality reduction, cluster cells, conduct differential AS analysis, and visualize splicing patterns. This protocol provides a reproducible and comprehensive framework for dissecting AS dynamics at single-cell resolution. It is readily adaptable to other SMART-Seq2 datasets and facilitates systematic investigation of splicing heterogeneity in diverse biological contexts.
Acute Contact and Oral Testing of Chemical Compounds on Vespa velutina nigrithorax (Hymenoptera, Vespidae) Under Laboratory Conditions

Standardized laboratory assays are essential for generating reproducible and comparable data in toxicology. Although acute contact and oral toxicity tests are widely applied in pesticide risk assessment, these approaches have rarely been adapted for social vespids. Vespa velutina nigrithorax, an invasive hornet in Europe and East Asia, is commonly managed through chemical control, yet treatment efficacy may vary depending on the route of exposure and other biological factors. This protocol describes a standardized method to assess acute contact and oral toxicity of chemical compounds in adult V. v. nigrithorax workers under controlled laboratory conditions. Hornets are collected in the field, individually housed, and exposed either to topical applications on the thorax or to spiked food sources. Mortality is monitored over 48–96 h and analyzed using appropriate statistical approaches to estimate lethal endpoints. This protocol enables comparison among compounds and exposure routes and provides a practical framework for toxicity screening in hornets.
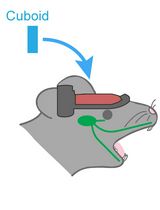
Hemispherectomy-Based Optical Window for In Vivo Visualization of Trigeminal Ganglion Neurons in Mice
Functional imaging of neural structures at the base of the cranium, including the trigeminal ganglion (TG), is technically challenging due to limited optical access. The TG—the largest sensory ganglion in the head—houses primary afferent neurons that relay information from the teeth, oral cavity, and face, yet investigation of somatosensory processing at the population level has remained limited. Here, we present a surgical procedure for an optical-window preparation that enables direct optical access to the TG. The ganglion is exposed by a large temporal craniotomy with removal of overlying tissue, and a glass cuboid is then placed in direct contact with the TG to suppress motion while maintaining the cranial cavity as a closed compartment without continuous perfusion. This preparation allows reliable visualization and recording of individual TG neurons during controlled stimulation of diverse facial and intraoral sites. Our approach provides a practical platform to map peripheral sensory representations within the TG and to investigate mechanisms underlying dental sensation, orofacial pain, and trigeminal circuit function.
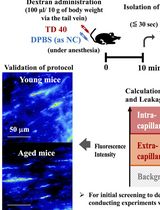
Quantitative Assessment of Capillary Permeability in Deep Intracardiac Capillaries Using Fluorescent Dextran
When the function of cardiac capillaries is impaired, cardiac function declines, and the risk of disease increases. No reliable assay has been developed to detect or evaluate the level of material exchange of capillaries deep within healthy heart tissue. In this study, we develop a new method to detect and evaluate molecules leaking from capillaries in cardiac tissue. By administering fluorescent dextran to mice via the tail vein, followed by rapid processing of the heart tissue, we have detected leaking fluorescent material from intracardiac microvessels. By comparing the detected images with those taken during the negative-control administration, using the image processing software LAS X and ImageJ, we detected trace amounts of fluorescent material that had leaked from the capillaries. We calculated the area of tissue where fluorescence was detected to perform a quantitative assessment, which we used as an indicator of capillary permeability. This new method of indexing will provide a different perspective on the factors contributing to the decline in cardiac function and the increased risk of disease with aging.
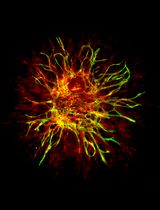
Stepwise Differentiation of Mouse Embryonic Stem Cells Into Murine Blood Vessel Organoids With Endothelial Lineage Tracing for Quality Control
In vitro vascular models are most informative when they recapitulate endothelial assembly within a 3D microenvironment. Blood vessel organoids (BVOs) enable the study of vascular heterogeneity, function, and organ-instructive cues in development, homeostasis, and disease. Here, we present a robust stepwise method to generate murine blood vessel organoids (mBVOs) from feeder-dependent mouse embryonic stem cells (mESCs) of common genetic backgrounds. Embryoid bodies (EBs) are formed using strain-specific seeding densities (day 0–3), followed by mesoderm induction (day 3–6) and vascular induction (day 6–8). Induced EBs are embedded in collagen I with Geltrex to drive sprouting and network formation (day 8–13). Vascular networks are microdissected and grown in suspension to yield mature mBVOs (day 21–30). The inclusion of a Cre-inducible VE-cadherin-GFP reporter line enables a quantitative quality control, reducing variability by excluding poorly differentiated organoids. The protocol reliably produces ~100 mBVOs per differentiation and is compatible with engineered mouse strains for gain- and loss-of-function studies, functional assays of vascular plasticity, and syngeneic grafting to assess perfusion. Thus, mBVOs provide a scalable and traceable 3D platform that bridges endothelial assays, mouse models, and human organoid systems.
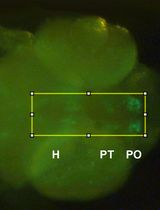
Whole-Mount Immunostaining of Tyrosine Hydroxylase for Dopaminergic Neuron Analysis in Zebrafish Larvae
Whole-mount techniques are widely used in medical and biological research to analyze protein expression and tissue organization in intact specimens. Traditional approaches for protein localization include section-based immunohistochemistry and in situ hybridization; however, these methods can be limited by tissue disruption and loss of spatial context. Whole-mount protocols generally involve tissue fixation, permeabilization, and staining with specific probes, but their effectiveness varies depending on the antigen–antibody combination and the specimen type. Consequently, no universal protocol is suitable for all experimental conditions. This protocol presents a detailed whole-mount immunostaining protocol for evaluating tyrosine hydroxylase (TH) expression, a key marker of dopaminergic neurons, in zebrafish (Danio rerio) larvae. The procedure outlines critical steps from sample preparation to staining optimization to ensure reproducible and specific signal detection. This approach enables accurate visualization and analysis of dopaminergic neuron distribution in intact larvae. The protocol offers a reliable and adaptable approach that preserves tissue integrity, enables three-dimensional visualization, and is particularly well-suited for developmental and neurobiological studies using zebrafish larvae.
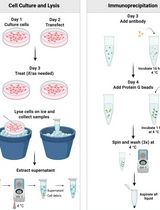
An Immunoprecipitation-Based Nonradioactive Kinase Assay to Measure Akt Kinase Activity in Mammalian Cell Lines
Protein kinase B, more commonly known as Akt, is a family of three serine/threonine kinases (Akt1, Akt2, and Akt3) that play a central role in regulating processes such as proliferation, survival, metabolism, and migration through phosphorylation of downstream targets. Given its involvement in numerous cellular processes, aberrant Akt signaling is prevalent across multiple cancer types, underscoring the need for Akt kinase assays to assess activity, regulatory mechanisms, and the efficacy of targeted interventions. Most existing Akt kinase assays rely on expensive commercial kits, some of which employ pre-purified, constitutively active Akt expressed in insect cells, bypassing physiologic autoinhibition of Akt; therefore, they are not suitable for evaluating allosteric inhibitors or context-dependent regulation. Here, we describe a detailed, step-by-step protocol for a nonradioactive Akt kinase assay using epitope-tagged, recombinant Akt1 expressed in a mammalian cell line and isolated by immunoprecipitation. This method eliminates the need to co-express Akt with upstream regulatory kinases or to purify active enzyme from insect cells, a time-consuming and technically demanding process, particularly when analyzing multiple Akt mutants. Because Akt is assayed in a regulated, autoinhibited state, this protocol enables direct evaluation of allosteric inhibitors that cannot be assessed using active Akt purified from insect cells. We note, however, that Akt1 kinase activity in this assay is measured from epitope-tagged, transiently overexpressed protein, which could influence cellular signaling dynamics. Despite this limitation, the cellular context preserves key regulatory features of Akt1 autoinhibition and membrane-dependent activation that are absent in assays using purified, pre-activated kinase. Together, this protocol supports analysis of Akt kinase activity under diverse experimental conditions, including receptor stimulation, pharmacologic treatment, allosteric inhibitor exposure, and mutations, using an accessible, economical, and physiologically relevant approach.
NADH-Dependent Oxidoreductase Activity Assay of OsAIM1 Using a Microplate Reader
Peroxisomal β-oxidation is a key step in jasmonic acid biosynthesis. Quantitative biochemical characterization of enzymes involved in the β-oxidation pathway is essential for validating their catalytic functions and comparing differences among genetic variants. Existing enzyme activity assays largely rely on chromatographic techniques to quantify substrate consumption or product formation, but these approaches are not well-suited for high-throughput or continuous kinetic measurements. Here, we describe a spectrophotometric assay based on a plate reader determining OsAIM1 enzymatic activity by monitoring the decrease in NADH absorbance at 340 nm. The method employs a 96-well plate reaction system, enabling real-time kinetic measurements and providing a standardized workflow for calculating reaction rates. Reaction components, protein concentration ranges, and data processing parameters were systematically optimized to ensure linearity, reproducibility, and quantitative accuracy. This assay is simple to perform, requires small reaction volumes, and offers relatively high throughput, making it suitable for functional characterization and kinetic analysis of NADH-dependent enzymes.
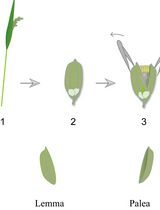
Lodicule Isolation and Morphometric Analysis During Rice Floret Opening
Rice lodicules are specialized floral organs located at the base of the ovary that undergo dynamic morphological changes during the flowering period. Water uptake–driven swelling and subsequent dehydration-induced shrinkage of the lodicules trigger floret opening and closure, respectively. Although lodicules play a central role in floret movement, standardized methods for quantitatively monitoring their temporal morphological changes remain limited. Here, we describe a detailed and reproducible workflow for lodicule sampling, dissection, imaging, and quantitative morphometric analysis. Florets are collected at predefined clock time points during the flowering period, and lodicules are carefully isolated under a stereomicroscope. High-resolution imaging is performed under consistent acquisition settings, followed by precise measurement of lodicule length, width, and thickness using image analysis software. This protocol emphasizes positional consistency in sampling, uniform imaging parameters, and standardized data analysis to enhance reproducibility. This method is suitable for evaluating the effects of genetic background or environmental conditions on lodicule morphology. By providing a standardized analytical framework, this protocol enables accurate and quantitative morphometric analysis of rice lodicules during floret opening.
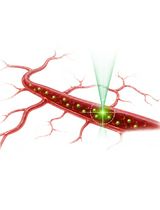
Protocol for In Vivo Two-Photon FCS to Measure Nanocarrier Number and Flow Velocity in Mouse Cerebral Microvasculature

Real-time measurement of blood flow and nanocarrier transport in the cerebral microvasculature is crucial for understanding neurovascular physiology and nanocarrier-based drug delivery. Existing techniques lack the ability to measure blood flow rates in individual vessels with high spatial and temporal resolution in real time. Two-photon fluorescence correlation spectroscopy (2P-FCS) provides a powerful approach for monitoring tracer molecules within a small confocal observation volume. This enables the simultaneous determination of particle number and flow dynamics in vivo. Here, we present a detailed protocol for in vivo 2P-FCS measurements in the mouse cerebral microvasculature. The protocol includes preparation of the cranial window, delivery of fluorescent dextran tracers for vascular visualization, and FCS measurements. It also includes two-photon imaging of the cerebrovascular network and acquisition and analysis of fluorescence correlation data. The protocol describes calibration of the confocal volume diameter and optimization of two-photon excitation parameters. This workflow enables real-time measurement of tracer concentration and flow velocity in individual cerebral microvessels with high spatial and temporal resolution. The method can be adapted to study blood flow dynamics, nanoparticle transport, and microvascular physiology in a variety of in vivo imaging systems equipped with multiphoton microscopy and FCS capabilities.
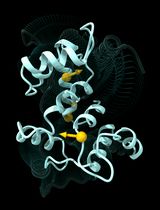
Multiply Perturbed Response: A Computational Protocol to Identify Cooperative Allosteric Residue Combinations Driving Protein Conformational Transitions

Construction and Functional Evaluation of Cyclic Peptide-Based CAR T Cells in Tumor Models
Cyclic peptides are emerging as a promising class of recognition modules for chimeric antigen receptor (CAR) engineering. Compared with single-chain variable fragment (scFv)-based CARs, disulfide-directed multicyclic peptides (DDMPs) represent a novel alternative, offering a markedly smaller molecular size (<5 kDa), enhanced structural stability through disulfide-directed cyclization, and broad tolerance to sequence diversification that supports systematic affinity and specificity optimization. DDMP-based CAR T cells leverage these properties to mediate antigen-dependent cytotoxicity while exhibiting an attenuated cytokine secretion profile, supporting the development of potentially safer immunotherapies for solid tumors. Here, we present a comprehensive workflow spanning CAR construct design and generation through in vitro and in vivo functional evaluation. While DDMPs are used as the exemplar recognition module, sections A and C–L of the protocol are directly applicable to any CAR format, including scFv- and nanobody-based designs with minimal modifications, making the workflow accessible to the broader CAR T-cell research community. The protocol includes the generation of Jurkat NFAT reporter cell lines and luciferase-expressing tumor target lines, which are widely used in different assays. Together, these standardized readouts enable rigorous, objective comparison of CAR T-cell efficacy and safety across tumor models.
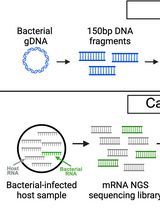
Enriching Bacteria-Specific RNA From Host Samples Before NGS With Transcript-Capture
Pathogen gene expression from host samples is often challenging to study due to low signal and high host RNA background. PCR probes have been recently used to hybridize and extract bacterial sequences from next-generation sequencing (NGS) libraries generated from in vitro and animal models of infection; however, these strategies require purchasing commercially synthesized probes that often do not capture the entire transcriptome. Transcript-capture sequencing is a novel capture approach for extracting RNA of a target bacterial species from samples in which there is substantial contamination by the host or other microbes. Biotinylated 150-base-pair DNA probes are generated in-house from bacterial DNA spanning the entire bacterial genome. Probes are hybridized to the cDNA of NGS sequencing libraries prepared from host samples to capture and enrich for bacterial-specific RNA reads before sequencing. This method results in a >200-fold increase in bacterial RNA reads from infected host samples (including in vitro, animal, and human samples) and generates complete bacterial transcriptomes with high gene coverage (>80%). Use of this protocol on infected host samples reveals a snapshot of bacterial activity during disease that may improve understanding of the physiological state of pathogens within their hosts.
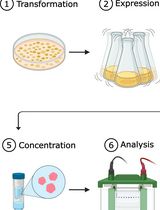
One-Step Affinity Purification of MarathonRT Reverse Transcriptase for RNA Sequencing Applications
Transfer RNAs (tRNAs) are important regulators of translation and cellular function. Several high-throughput sequencing methods have been developed to quantitatively analyze tRNA isoacceptors in cells. However, the strong secondary structures and extensive post-transcriptional modification of most tRNA molecules present significant challenges for many reverse transcriptases, negatively impacting sequencing library preparation and causing quantification biases. Currently, the field utilizes processive next-generation reverse transcriptases (ngRTs), such as Induro (New England Biolabs) and UltraMarathonRT (RNAConnect), to address these issues. Despite being used in multiple protocols, these commercial products face little competition and remain costly. However, non-commercial alternatives, such as the original MarathonRT (MRT), are available from gene repositories. MRT is a next-generation reverse transcriptase derived from the Eubacterium rectale group II intron maturase, which can read through RNA secondary structures and chemical modifications. Here, we present a simplified expression and purification protocol for producing highly active MRT that is stable over 1 year. This cost-effective protocol yields a heterogeneous protein preparation with no discernible competing enzymatic activities; it mitigates previously reported precipitation issues, saving one day of laboratory work and eliminating two chromatography-based purification steps. Moreover, the use of the resulting protein preparation has been verified in the mim-tRNAseq pipeline, where it was shown to perform equally to the commercial alternatives Induro and UltraMarathonRT. In addition, we have developed a simple and cost-effective assay for measuring the enzymatic activity of MRT, allowing for batch comparison.
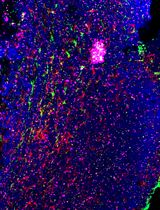
Simultaneous Immunofluorescence-Based In Situ mRNA Expression and Protein Detection in Bone Marrow Biopsy Samples
Fluorescence in situ hybridization (FISH) can be employed to study the expression and subcellular localization of nucleic acids by using labeled antisense strands that hybridize with the target RNA or DNA molecules. Likewise, immunofluorescence antibody staining (IF) takes advantage of the specific interaction between a fluorophore-labeled antibody and its corresponding antigen. This protocol reports the combination of RNA-FISH and IF antibody staining for simultaneous detection of both RNA transcripts and proteins of interest in routine formalin-fixed paraffin-embedded (FFPE) bone marrow biopsy samples. Herein, we provide a detailed description of the methodology that we have developed and optimized to study the spatial expression of two transcripts—TGFB1 and PDGFA1—in human hematopoietic (CD45+) and non-hematopoietic (CD271+) cells in the bone marrow of patients with acute lymphoblastic leukemia (ALL).
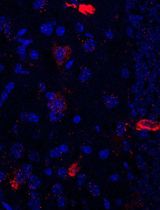
Using combined fluorescent in situ hybridization with Immunohistochemistry to co-localize mRNA in diverse neuronal cell types
Understanding gene expression within defined neuronal populations is essential for dissecting the cellular and molecular diversity of the brain. mRNA assays provide a direct readout of gene expression, capturing transcriptional changes that may precede or occur independently of protein abundance, whereas protein assays reflect the cumulative effects of translation, modification, and degradation. Moreover, in histological analysis, immunohistochemical protein detection results in visually diffuse labeling, which makes it difficult to quantitatively assess levels and locations of expression at high resolution. Here, we present a protocol that allows for mRNA detection in single neuronal cell types with a high degree of sensitivity and anatomical resolution. This protocol combines fluorescent in situ hybridization (FISH) with immunohistochemistry (IHC) on the same tissue section. Briefly, FISH is carried out by ACDBio RNAscope® fluorescent in situ hybridization technology, which involves processing the tissue sections, followed by signal amplification. This involves target retrieval, probe hybridization, and signal enhancement. Then, the tissue section is processed for IHC, which involves blocking nonspecific sites and incubation with primary antibodies, followed by development of a fluorescent signal with secondary antibodies. Typically, visual mRNA detection with FISH can be seen as individual puncta, whereas targeting the protein with an antibody results in filled cells or processes. The variation in staining pattern allows for the quantification of distinct mRNA transcripts within different neuronal populations, which renders co-localization analyses easy and efficient.

Electrophoretic Mobility Shift Assay (EMSA) for Assessing RNA–Protein Binding and Complex Formation Using Recombinant RNA-Binding Proteins and In Vitro–Transcribed RNA

Evaluating RNA–protein interactions is key to understanding post-transcriptional gene regulation. Electrophoretic mobility shift assays (EMSAs) remain a widely used technique to study these interactions, revealing information about binding affinities and binding modalities, including cooperativity and complex formation. Here, we detail, in a step-by-step protocol, how to perform EMSAs. We describe how to generate, purify, and quantitate 32P-radiolabeled RNA by in vitro transcription, as well as the expression and purification of recombinant RNA-binding proteins in E. coli using ELAV as an example. We then describe how to set up binding reactions using serial dilutions in a microtiter plate format of recombinant ELAV and in vitro–transcribed RNA and how to perform EMSAs using native low-crosslinked acrylamide gels, with detailed graphically supported instructions and troubleshooting guides.
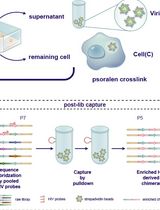
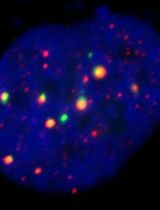
High-resolution mapping of RNA-RNA interactions across the HIV-1 genome with HicapR
基于 HiCapR 的 HIV-1 全基因组 RNA–RNA 相互作用高分辨率图谱构建
The genomes of RNA viruses can fold into dynamic structures that regulate their own infection and immune evasion processes. Proximity ligation methods (e.g., SPLASH) enable genome-wide interaction mapping but lack specificity when dealing with low-abundance targets in complex samples. Here, we describe HiCapR, a protocol integrating in vivo psoralen crosslinking, RNA fragmentation, proximity ligation, and hybridization capture to specifically enrich viral RNA–RNA interactions. Captured libraries are sequenced, and chimeric reads are analyzed via a customized computational pipeline to generate constrained secondary structures. HiCapR generates high-resolution RNA interaction maps for viral genomes. We applied it to resolve the in vivo structure of the complete HIV-1 RNA genome, identifying functional domains, homodimers, and long-range interactions. The protocol's robustness has been previously validated on the SARS-CoV-2 genome. HiCapR combines proximity ligation with targeted enrichment, providing an efficient and specific tool for studying RNA architecture in viruses, with broad applications in virology and antiviral development.

Enhanced RNA-Seq Expression Profiling and Functional Enrichment in Non-model Organisms Using Custom Annotations
Functional enrichment analysis is essential for understanding the biological significance of differentially expressed genes. Commonly used tools such as g:Profiler, DAVID, and GOrilla are effective when applied to well-annotated model organisms. However, for non-model organisms, particularly for bacteria and other microorganisms, curated functional annotations are often scarce. In such cases, researchers often rely on homology-based approaches, using tools like BLAST to transfer annotations from closely related species. Although this strategy can yield some insights, it often introduces annotation errors and overlooks unique species-specific functions. To address this limitation, we present a user-friendly and adaptable method for creating custom annotation R packages using genomic data retrieved from NCBI. These packages can be directly imported as libraries into the R environment and are compatible with the clusterProfiler package, enabling effective gene ontology and pathway enrichment analysis. We demonstrate this approach by constructing an R annotation package for Mycobacterium tuberculosis H37Rv, as an example. The annotation package is then utilized to analyze differentially expressed genes from a subset of RNA-seq dataset (GSE292409), which investigates the transcriptional response of M. tuberculosis H37Rv to rifampicin treatment. The chosen dataset includes six samples, with three serving as untreated controls and three exposed to rifampicin for 1 h. Further, enrichment analysis was performed on genes to demonstrate changes in response to the treatment. This workflow provides a reliable and scalable solution for functional enrichment analysis in organisms with limited annotation resources. It also enhances the accuracy and biological relevance of gene expression interpretation in microbial genomics research.
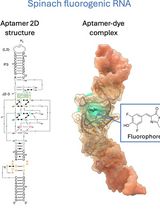
Visualizing diverse RNA functions in living cells with Spinach™ family of fluorogenic aptamers
利用SpinachTM系列荧光适配体可视化活细胞中多种RNA功能
RNA is now recognized as a highly diverse and dynamic class of molecules whose localization, processing, and turnover are central to cell function and disease. Live-cell RNA imaging is therefore essential for linking RNA behavior to mechanism. Existing approaches include quenched hybridization probes that directly target endogenous transcripts but face delivery and sequestration issues, protein-recruitment tags such as MS2/PP7 that add large payloads and can perturb localization or decay, and CRISPR–dCas13 imaging that requires substantial protein cargo and careful control of background and off-target effects. Here, we present a protocol for live-cell RNA imaging using the SpinachTM family of fluorogenic RNA aptamers. The method details the design and cloning of SpinachTM-tagged RNA constructs, selection and handling of cognate small-molecule fluorophores, expression in mammalian cell lines, dye loading, and image acquisition on standard fluorescence microscopes, followed by quantitative analysis of localization and dynamics. We include controls to verify aptamer expression and signal specificity, guidance for multiplexing with related variants (e.g., Broccoli, Corn, Squash, Beetroot), and troubleshooting for dye permeability and signal optimization. Application examples illustrate use in tracking cellular delivery of mRNA therapeutics, monitoring transcription and decay in response to perturbations, and the forming of toxic RNA aggregates. Compared with prior methods, SpinachTM tags are compact, genetically encodable, and fluorogenic, providing high-contrast imaging in both the nucleus and cytoplasm with single-vector simplicity and multiplexing capability. The protocol standardizes key steps to improve robustness and reproducibility across cell types and laboratories.
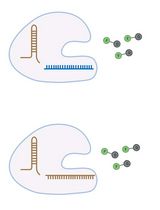
Amplification-Free Detection of Highly Structured RNA Molecules Using SCas12aV2
The CRISPR/Cas12a system has revolutionized molecular diagnostics; however, conventional Cas12a-based methods for RNA detection typically require transcription and pre-amplification steps. Our group has recently developed a diagnostic technique known as the SCas12a assay, which combines Cas12a with a split crRNA, achieving amplification-free detection of miRNA. However, this method still encounters challenges in accurately quantifying long RNA molecules with complex secondary structures. Here, we report an enhanced version termed SCas12aV2 (split-crRNA Cas12a version 2 system), which enables direct detection of RNA molecules without sequence limitation while demonstrating high specificity in single-nucleotide polymorphism (SNP) applications. We describe the general procedure for preparing the SCas12a system and its application in detecting RNA targets from clinical samples.
Enhancement of RNA Imaging Platforms by the Use of Peptide Nucleic Acid-Based Linkers
RNA imaging techniques enable researchers to monitor RNA localization, dynamics, and regulation in live or fixed cells. While the MS2-MCP system—comprising the MS2 RNA hairpin and its binding partner, the MS2 coat protein (MCP)—remains the most widely used approach, it relies on a tag containing multiple fluorescent proteins and has several limitations, including the potential to perturb RNA function due to the tag’s large mass. Alternative methods using small-molecule binding aptamers have been developed to address these challenges. This protocol describes the synthesis and characterization of RNA-targeting probes incorporating a peptide nucleic acid (PNA)-based linker within the cobalamin (Cbl)-based probe of the Riboglow platform. Characterization in vitro involves a fluorescence turn-on assay to determine binding affinity (KD) and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) footprinting analysis to assess RNA-probe interactions at a single nucleotide resolution. To show the advancement of PNA probes in live cells, we present a detailed approach to perform both stress granule (SG) and U-body assays. By combining sequence-specific hybridization with structure-based recognition, our approach enhances probe affinity and specificity while minimizing disruption to native RNA behavior, offering a robust alternative to protein-based RNA imaging systems.






