

A Bioinformatics Workflow to Identify eccDNA Using ECCFP From Long-Read Nanopore Sequencing Data
(*contributed equally to this work) 发布: 2026年03月20日第16卷第6期 DOI: 10.21769/BioProtoc.5636 浏览次数: 15
评审: Migla MiskinyteYuhang WangAnonymous reviewer(s)








Abstract
Extrachromosomal circular DNA (eccDNA) is a type of circular DNA that exists independently of chromosomes and has garnered significant attention in various fields, particularly in the context of smaller eccDNAs, which have considerable roles in gene regulation through various mechanisms. Current methods such as Circle-Seq and 3SEP can enrich small eccDNAs during sample preparation, but most bioinformatics pipelines remain challenging, exhibiting low accuracy and efficiency. This protocol describes the detailed workflow of a newly developed bioinformatics analysis pipeline, named EccDNA Caller based on Consecutive Full Pass (ECCFP), to accurately identify eccDNA from long-read Nanopore sequencing data. Compared to other pipelines, ECCFP significantly improves detection sensitivity, accuracy, and runtime efficiency. The process includes raw data quality control, trimming of adapters and barcodes, alignment to a reference genome, and identification of eccDNA, with detailed results encompassing accurate positioning of eccDNA, consensus sequences, and variants of individual eccDNA.
Key features
• This protocol provides a beginner-friendly, step-by-step workflow that enables researchers without bioinformatics experience to successfully execute the entire eccDNA identification process.
• It offers an efficient computational pipeline for eccDNA detection from Nanopore sequencing data, integrating quality control, trimming, alignment, and eccDNA identification.
• ECCFP exhibits sensitivity, accuracy, high efficiency, and low false-positive rates compared to existing long-read-based tools.
Keywords: ECCFPGraphical overview
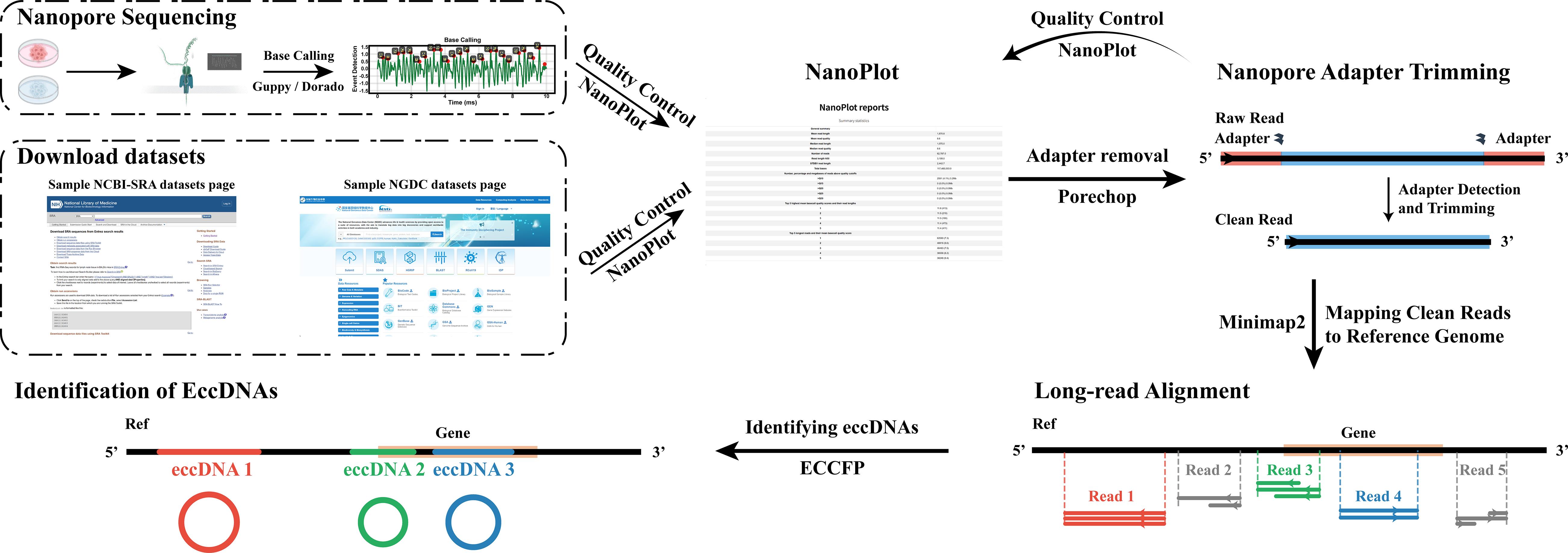
The entire process from Nanopore sequencing data acquisition to eccDNA identification
Background
Extrachromosomal circular DNA (eccDNA) is a type of genetic material that exists independently of chromosomes and is widely found in eukaryotic organisms [1,2]. Recent studies have highlighted its significant role in various biological functions, particularly in cancer initiation, progression, and drug resistance [3–6]. In addition, eccDNA varies from tens to millions of base pairs (bp) in length, where molecules exceeding 10 kb are classified as ecDNA [7]. These large circular DNA molecules have been identified in over half of all human cancers, primarily using FISH and whole-genome sequencing (WGS) for validation [8,9]. Current experimental methods developed for the enrichment of small eccDNA, such as Circle-Seq [10] and 3SEP [11,12], typically identify eccDNA molecules smaller than 10 kb. These smaller eccDNA molecules have been demonstrated to regulate gene expression through various mechanisms [13].
Both Circle-Seq and 3SEP experimental methodologies utilize rolling circle amplification (RCA). During RCA, DNA polymerase continually traverses circular templates, producing concatemeric tandem copies (CTCs) [14]. Currently, most bioinformatics pipelines for eccDNA detection are based on overly strict selection criteria for CTCs [15] and often overlook the complexities introduced by rolling circle amplification (RCA) and random errors during the sequencing process, which might result in ghost sequences.
Recently, our group developed a novel bioinformatics pipeline, named EccDNA Caller based on Consecutive Full Pass (ECCFP) [16], for eccDNA detection from long-read Nanopore sequencing data. ECCFP utilizes all individual sequencing reads, including ghost and non-ghost sequences, to identify candidate eccDNA molecules. The pipeline then integrates these candidate eccDNAs and employs the Boyer–Moore majority vote algorithm to accurately determine eccDNA positions and gather information on circular consensus sequences. Compared to other current pipelines that identify small eccDNA molecules from long-read sequencing data using RCA amplification, ECCFP combines the strengths of these pipelines and is optimized specifically for RCA data, resulting in improved sensitivity, accuracy, and operational efficiency.
Briefly, the current signal data from Nanopore sequencing, typically stored in FAST5 or POD5 formats, undergo base calling through tools such as Guppy or Dorado. This computational conversion transforms the current signal data into nucleotide sequence data, ultimately producing demultiplexed FASTQ files for downstream genomic analyses. Upon FASTQ acquisition, initial quality assessment is performed using NanoPlot. Adapter and barcode sequences are then removed with Porechop, followed by post-trimming quality re-evaluation of trimmed reads using NanoPlot. Trimmed reads require alignment to a reference genome for sequence mapping. Minimap2 serves as the optimal aligner for long-read data due to its efficient handling of error-prone sequences. Subsequent eccDNA detection is conducted using ECCFP, which identifies circular DNA through structural signature analysis of aligned reads. The entire process from Nanopore sequencing data acquisition to eccDNA identification can be referenced to in the Graphical Overview for clearer understanding. This protocol details the core bioinformatic workflow for eccDNA detection, encompassing quality control, adapter and barcode trimming, reference genome alignment, and circular DNA identification. As a computational pipeline, it exclusively addresses analytical methodologies while explicitly excluding wet laboratory procedures. All sequencing data used are derived from public repositories, including Project PRJCA040952 and PRJCA052047 deposited by our group at NGDC alongside datasets from published studies.
This protocol relies exclusively on open-source software tools that are readily accessible. Furthermore, we provide comprehensively documented analytical code designed to guide users through the complete workflow from FASTQ processing to eccDNA identification. This makes the protocol accessible even for researchers without prior bioinformatics experience.
Software and datasets
Hardware
The analysis was performed on a Linux/Unix system in the following benchmarked configuration:
1. Memory (RAM): 512 GB
2. CPU: 64-bit architecture, 18 cores, 72 threads
Note: This setup demonstrates robust performance for the entire workflow. Hardware specifications can be adjusted based on data scale and resource availability.
Software
1. Conda (version 24.9.2, https://anaconda.org/anaconda/conda)
2. Python (version 3.12.2, https://www.python.org/downloads/)
3. ECCFP (version 1.0.1, https://github.com/WSG-Lab/ECCFP, MIT License)
4. Porechop (version 0.2.4, https://github.com/rrwick/Porechop, GNU General Public License v3.0)
5. Minimap2 (version 2.28, https://github.com/lh3/minimap2, MIT License) [17,18]
6. NanoPlot (version 1.42.0, https://github.com/wdecoster/NanoPlot, MIT License) [19]
Note: Conda is used as the package manager and Python as the interpreter. Although there are no absolute version requirements, to ensure reproducibility of the environment and avoid compatibility issues, we strongly recommend using the above suggested version combinations.
Software requirements: dependencies
1. numpy (version 1.26.4, https://numpy.org/)
2. pandas (version 2.3.2, https://pandas.pydata.org/ )
3. rich (version 14.1.0, https://rich.readthedocs.io/ )
4. biopython (version 1.85, https://biopython.org/)
5. pyfaidx (version 0.9.0.1, https://github.com/mdshw5/pyfaidx)
6. pyfastx (version 2.2.0, https://github.com/lmdu/pyfastx) [20]
Note: Although specific versions are listed for completeness, only minimap2, numpy, pandas, and biopython have strict version requirements. While version inconsistencies in other tools are unlikely to critically impact results, we strongly recommend maintaining a consistent software environment to ensure full reproducibility and computational stability.
Data
All sequencing data generated and analyzed in this study are publicly accessible through the National Genomics Data Center (NGDC) and the NCBI Sequence Read Archive (SRA). Specifically, the NGDC hosts data under project accessions PRJCA040952 and PRJCA052047 (generated and deposited by our laboratory; access requires an application to the database) as well as PRJCA010264. Detailed application guidelines for PRJCA040952 are provided in the official documentation ( https://ngdc.cncb.ac.cn/gsa-human/document/GSA-Human_Request_Guide_for_Users_us.pdf) and are not reiterated here. Data are available from the NCBI SRA under project accession PRJNA806866, from which the following four sample IDs were utilized: SRR18143375, SRR18143376, SRR18143377, and SRR18143378 (Table 1). All analyzed samples originated from human cell lines. Genomic alignments and subsequent analyses were based on the GRCh38.p14 (hg38) reference genome assembly obtained from the GENCODE database.
Table 1. Summary of publicly available sequencing datasets
| Project | Data_id | Sample |
|---|---|---|
| PRJCA040952 | HRR2590080 | HepG2 cell |
| PRJCA010264 | HRR695439 | BGC823 cell |
| PRJCA010264 | HRR695440 | SGC7901 cell |
| PRJCA010264 | HRR695441 | GES1 cell |
| PRJCA010264 | HRR695442 | HepG2 cell |
| PRJCA010264 | HRR695443 | HL7702 cell |
| PRJCA010264 | HRR695444 | MDA-MB-453 cell |
| PRJCA010264 | HRR695445 | MCF12A cell |
| PRJNA806866 | SRR18143375 | EJM cells |
| PRJNA806866 | SRR18143376 | JJN3 cell |
| PRJNA806866 | SRR18143377 | APR1 cell |
| PRJNA806866 | SRR18143378 | APR1 cell |
Procedure
文章信息
稿件历史记录
提交日期: Dec 10, 2025
接收日期: Feb 9, 2026
在线发布日期: Feb 27, 2026
出版日期: Mar 20, 2026
版权信息
© 2026 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Li, W., Miao, B. and Wan, S. (2026). A Bioinformatics Workflow to Identify eccDNA Using ECCFP From Long-Read Nanopore Sequencing Data. Bio-protocol 16(6): e5636. DOI: 10.21769/BioProtoc.5636.
分类
生物信息学与计算生物学
生信
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。