

Identifying Causal Genes and Building Regulatory Networks in Crops Using the CisTrans-ECAS Method
基于 CisTrans-ECAS 方法的作物因果基因鉴定与调控网络构建
发布: 2026年02月05日第16卷第3期 DOI: 10.21769/BioProtoc.5578 浏览次数: 151
评审: Anonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Nov 2023









Abstract
Pinpointing causal genes for complex traits from genome-wide association studies (GWAS) remains a central challenge in crop genetics, particularly in species with extensive linkage disequilibrium (LD) such as rice. Here, we present CisTrans-ECAS, a computational protocol that overcomes this limitation by integrating population genomics and transcriptomics. The method’s core principle is the decomposition of gene expression into two distinct components: a cis-expression component (cis-EC), regulated by local genetic variants, and a trans-expression component (trans-EC), influenced by distal genetic factors. By testing the association of both components with a phenotype, CisTrans-ECAS establishes a dual-evidence framework that substantially improves the reliability of causal inference. This protocol details the complete workflow, demonstrating its power not only to identify causal genes at loci with weak GWAS signals but also to systematically reconstruct gene regulatory networks. It provides a robust and powerful tool for advancing crop functional genomics and molecular breeding.
Key features
• Pinpointing causal genes with high precision: Integrates cis- and trans-expression components to distinguish true causal genes from LD artifacts, even for small-effect loci.
• Reconstructing gene regulatory networks: Uses gene expression as molecular traits to identify upstream regulators, revealing complex molecular regulatory pathways.
• Versatile and reproducible workflow: An R-based pipeline using PLINK and GCTA, applicable to rice and other species with population genomics and transcriptomics data.
• Experimentally validated reliability: The method successfully identified key genes OsMADS17 and SDT that regulate rice spikelet number, with their regulatory relationship confirmed by molecular experiments.
Keywords: Causal gene identification (因果基因鉴定)Graphical overview
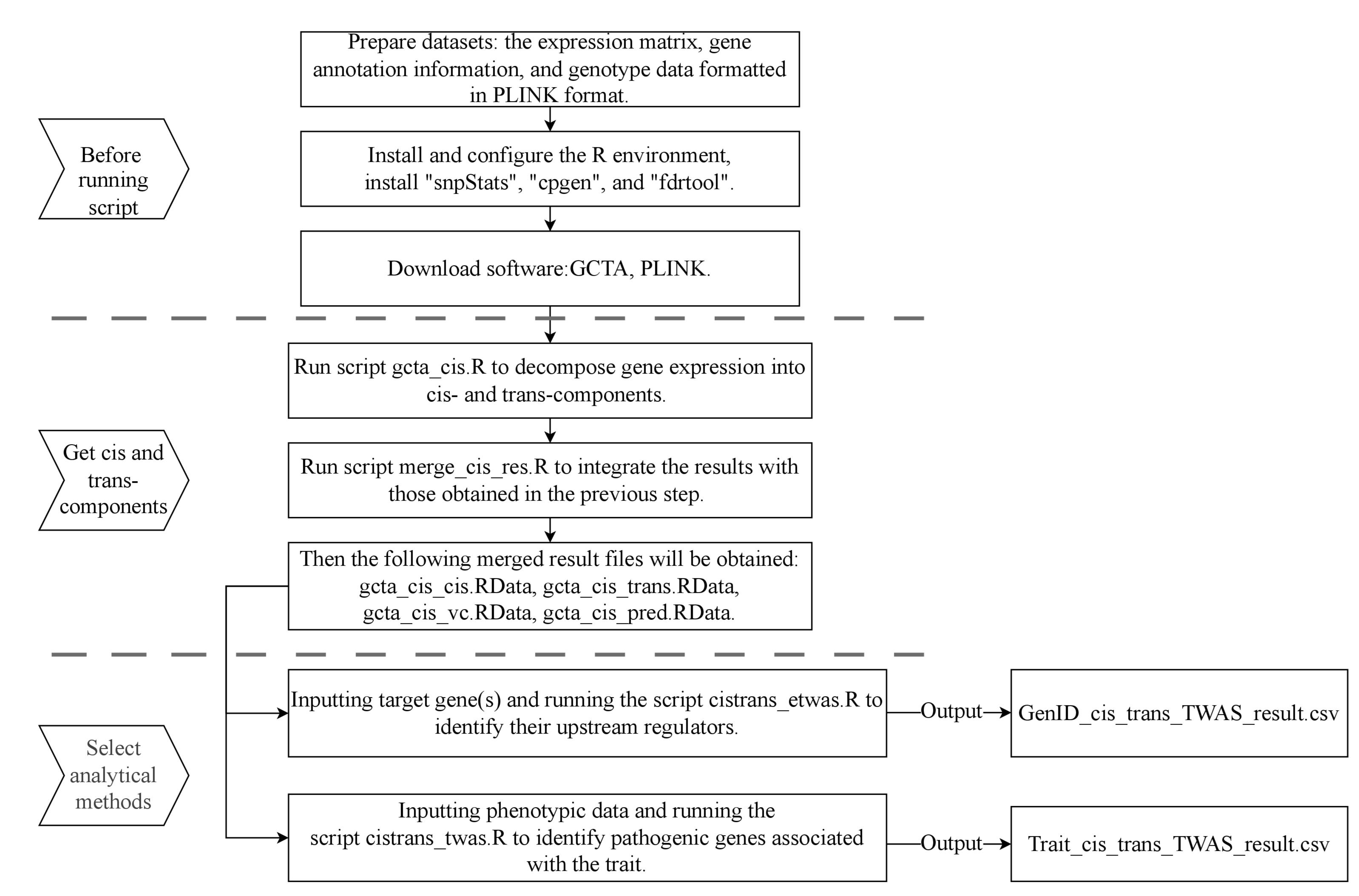
Flowchart of the CisTrans-ECAS method. The workflow is divided into three main stages: preparation before running scripts, the process of obtaining cis-EC and trans-EC, and the selection of analytical methods for downstream analysis. The flowchart details the key scripts (gcta_cis.R, merge_cis_res.R, cistrans_etwas.R, cistrans_twas.R) and their respective inputs and outputs.
Background
Important agronomic traits in crops, such as yield, quality, and stress resistance, are typically complex traits controlled by multiple genes. Genome-wide association studies (GWAS) are a primary tool for dissecting the genetic basis of these traits. However, in many crops like rice, extensive linkage disequilibrium (LD) and the polygenic nature of traits mean that a single GWAS locus can harbor dozens of genes, making the identification of the true causal gene a formidable challenge. This significantly limits the practical application of GWAS findings.
The advent of transcriptomics has opened new avenues to tackle this issue. Gene expression, as an intermediate layer between genotype and phenotype, is itself under genetic control. Methods like expression quantitative trait loci (eQTL) analysis and TWAS link genetic variants to gene expression levels, providing functional evidence to prioritize candidate genes. Recent TWAS frameworks (e.g., PrediXcan- and FUSION-based approaches) and eQTL-guided colocalization analyses provide functional evidence to link genes with complex traits and have been applied in both human and plant genetics [1–3]. These models predict gene expression from the genotype and test its association with phenotypes, offering a mechanistic layer beyond SNP-trait correlations. Colocalization-based methods further evaluate whether GWAS and eQTL signals share a common causal variant, helping prioritize functional loci. While these approaches have proven valuable, their performance in crops is often hindered by extensive LD, complex population structure, and the difficulty of distinguishing traits driven by local regulation from those influenced by distal regulatory networks.
To address this challenge, we developed the CisTrans-ECAS (cis- and trans-expression component-based association study) method. Its core concept is that the expression variation of a gene can be partitioned into two components: 1) the cis-expression component (cis-EC), in which expression variation is explained by local genetic variants (e.g., within 100 kb upstream and downstream); and 2) the trans-expression component (trans-EC), in which expression variation is driven by distal regulatory factors or other genetic influences not captured by the cis-EC.
Unlike traditional TWAS methods that rely on total gene expression or predicted expression levels, CisTrans-ECAS explicitly decomposes expression into cis- and trans-regulated components. This separation allows the method to distinguish between local regulatory effects and broader regulatory network influences, providing higher specificity for identifying causal genes in regions of extensive LD. Our rationale is that if a gene is a true functional participant in a biological process affecting a trait, its association with the trait should be evident from two independent sources. Its local regulation (cis-EC) should be linked to the trait, and its regulation within a broader network (trans-EC) should also be associated with the trait.
Therefore, a gene whose cis-EC and trans-EC are both significantly associated with a target trait is a much stronger causal candidate than a gene with only one type of association. This dual-evidence strategy effectively filters out false positives arising from LD, enabling the precise identification of causal genes, even in regions with weak GWAS signals. Furthermore, by treating the expression of other genes as molecular phenotypes (e-traits), this method can efficiently map upstream and downstream regulatory relationships, providing robust support for establishing reliable gene regulatory networks.
Software and datasets
A. Install required software
This protocol was tested in a Linux server environment. The core analyses rely on PLINK, GCTA, and several R packages (Table 1). While originally developed using R v3.5.1, the protocol is expected to be compatible with recent versions (e.g., R 4.x). We recommend creating a dedicated Conda environment.
1. Create and activate a Conda environment:
# Create and activate a conda environmentconda create -n ecas_env r-base=3.5.1conda activate ecas_env2. Install R packages:
Within the activated conda environment, start R and run the following commands to install the required packages.
# Install from CRANinstall.packages("fdrtool")# Install from condaconda install bioconda::bioconductor-snpstatsconda install -c conda-forge r-devtoolsconda install -c conda-forge r-lme4# Install from GitHubdevtools::install_github("cheuerde/cpgen", ref = "master", build_vignettes=FALSE)Table 1. Software and resources for data analysis.
| Type | Software/dataset/resource | Version | OS | License | Access/source |
|---|---|---|---|---|---|
| Software | PLINK | 1.9 | Linux | GPLv2 | https://www.cog-genomics.org/plink/ |
| Software | GCTA | 1.93.2beta | Linux | GPLv3 | https://yanglab.westlake.edu.cn/software/gcta/ |
| Software | snpStats | 1.32.0 | R | GPLv2 | Bioconductor |
| Software | cpgen | 0.2 | R | GPLv3 | GitHub |
| Software | fdrtool | 1.2.16 | R | GPLv2 | CRAN |
B. Companion GitHub repository for protocol implementation
All scripts, example data, and detailed instructions required for this protocol are available in the following GitHub repository:
• Repository: https://github.com/Minglc/CisTrans-ECAS
• DOI for the code: https://doi.org/10.5281/zenodo.10004834
It is highly recommended to read the README.md file carefully before starting.
Procedure
文章信息
稿件历史记录
提交日期: Oct 8, 2025
接收日期: Dec 17, 2025
在线发布日期: Jan 5, 2026
出版日期: Feb 5, 2026
版权信息
© 2026 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Yan, Y., Ming, L. and Xie, W. (2026). Identifying Causal Genes and Building Regulatory Networks in Crops Using the CisTrans-ECAS Method. Bio-protocol 16(3): e5578. DOI: 10.21769/BioProtoc.5578.
分类
生物信息学与计算生物学
系统生物学 > 相互作用组 > 基因网络
植物科学 > 植物分子生物学 > 遗传分析
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。