

现期刊物2026
卷册: 16, 期号: 6

生物信息学与计算生物学

A Bioinformatics Workflow to Identify eccDNA Using ECCFP From Long-Read Nanopore Sequencing Data
利用 ECCFP 从 Nanopore 长读长测序数据中鉴定细胞外染色体环状 DNA 的生物信息学流程
癌症生物学

Mag-Net Strong Anion Exchange Enables Isolation of Ovarian Cancer Ascites Extracellular Vesicles for Proteomic Biomarker Discovery
Mag-Net 强阴离子交换技术用于分离卵巢癌腹水来源细胞外囊泡以开展蛋白质组学标志物发现
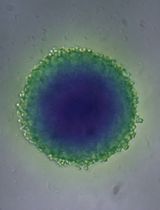
A Simple and Cost-Effective Method for Generating Spheroids From Triple-Negative Breast Cancer Cell Line (MDA-MB-231)
一种简单且低成本的三阴性乳腺癌细胞系(MDA-MB-231)球状体构建方法


细胞生物学
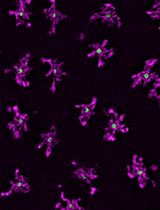
Radial Profile-Based Quantification of Centrosomal Proteins
基于径向分布分析的中心体蛋白定量方法
微生物学
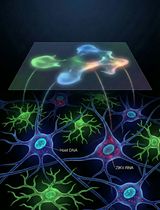
A Novel Sequencing Method for Quantification of ZIKV RNA in Individual Cells
一种用于单细胞水平定量寨卡病毒RNA的新型测序方法
分子生物学
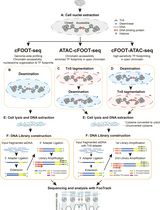
A Cytosine Deaminase–Based Genomic Footprinting Assay (cFOOT-seq) for Detecting Transcription Factor Occupancy
基于胞嘧啶脱氨酶的基因组足迹分析方法(cFOOT-seq)用于检测转录因子占据情况
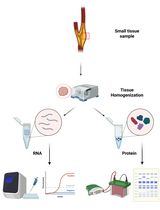
Tandem RNA and Protein Extraction: A Platform for Maximizing the Use of Limited Ex Vivo Tissue Samples
RNA与蛋白质串联提取方法:最大化利用有限离体组织样本的实验平台
植物科学
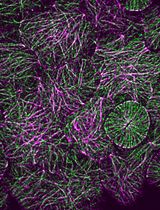
A Guide to Reproducible Cellulose Synthase Density and Speed Measurements in Arabidopsis thaliana
拟南芥中纤维素合酶密度与运动速度测量的可重复性分析指南
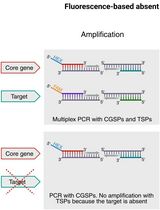
Fluorescence-Based Absent Allele-Specific Amplification (FAASA) for High-Throughput Detection of Absent Alleles
基于荧光的缺失等位基因特异性扩增(FAASA):用于高通量检测缺失等位基因的方法

A Rapid and Visual Soybean Hairy Root Transformation Protocol Using the RUBY Reporter
利用RUBY可视化报告系统的快速大豆毛状根转化方法

Controlled Transmission of a Fijivirus Under Field Conditions Using Mass-Reared Planthoppers
利用规模化饲养飞虱在田间条件下实现 Fijivirus 的可控传播方法